

The MEK/ERK Module Is Reprogrammed in Remodeling Adult Cardiomyocytes
- PMID: 32882982
- PMCID: PMC7503571
- DOI: 10.3390/ijms21176348
The MEK/ERK Module Is Reprogrammed in Remodeling Adult Cardiomyocytes
Abstract
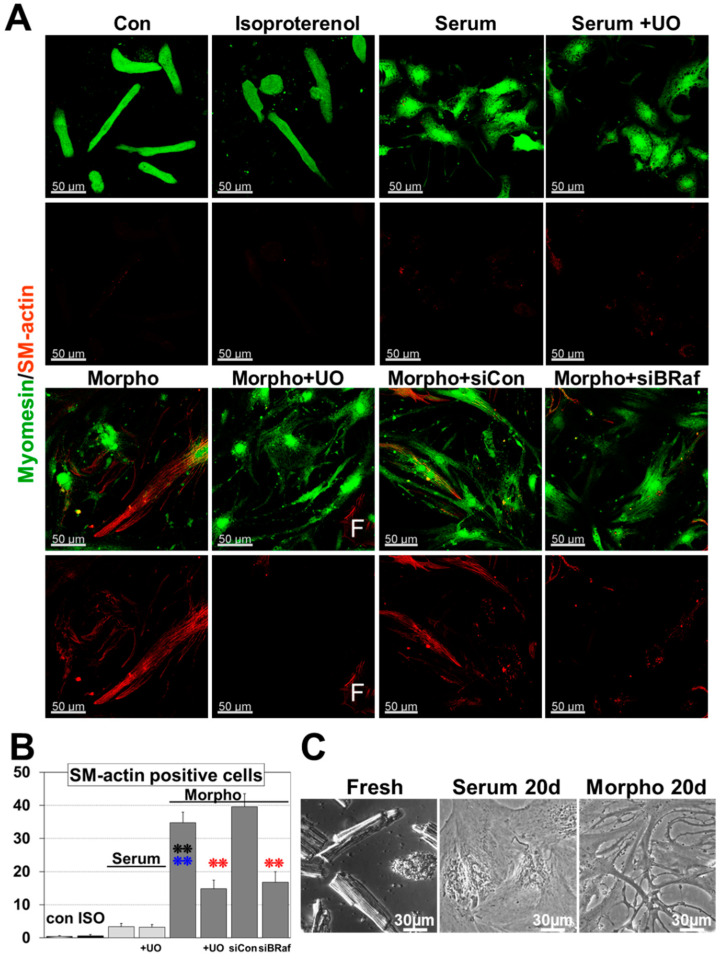
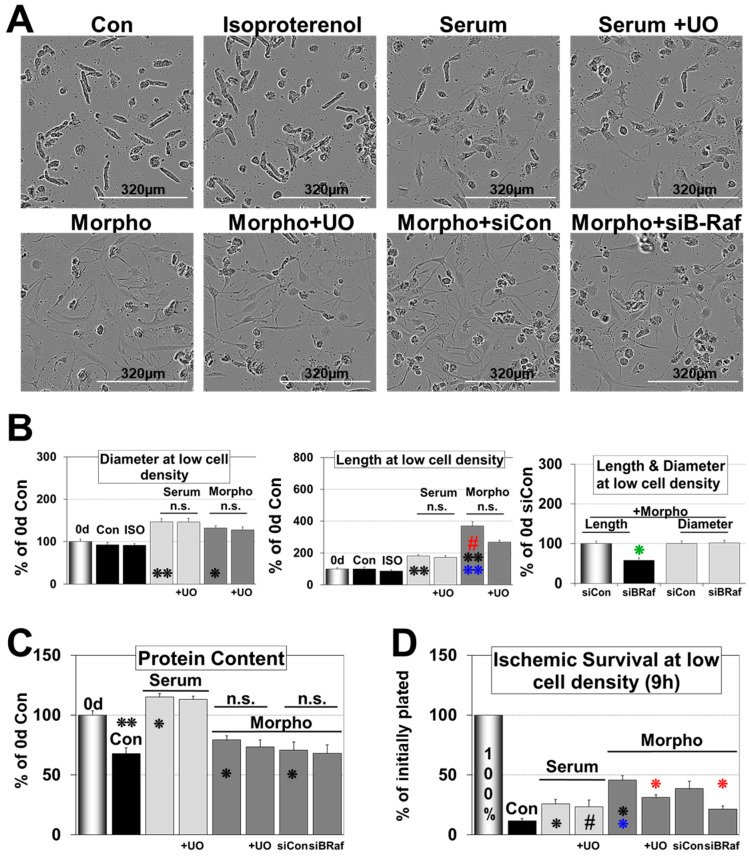
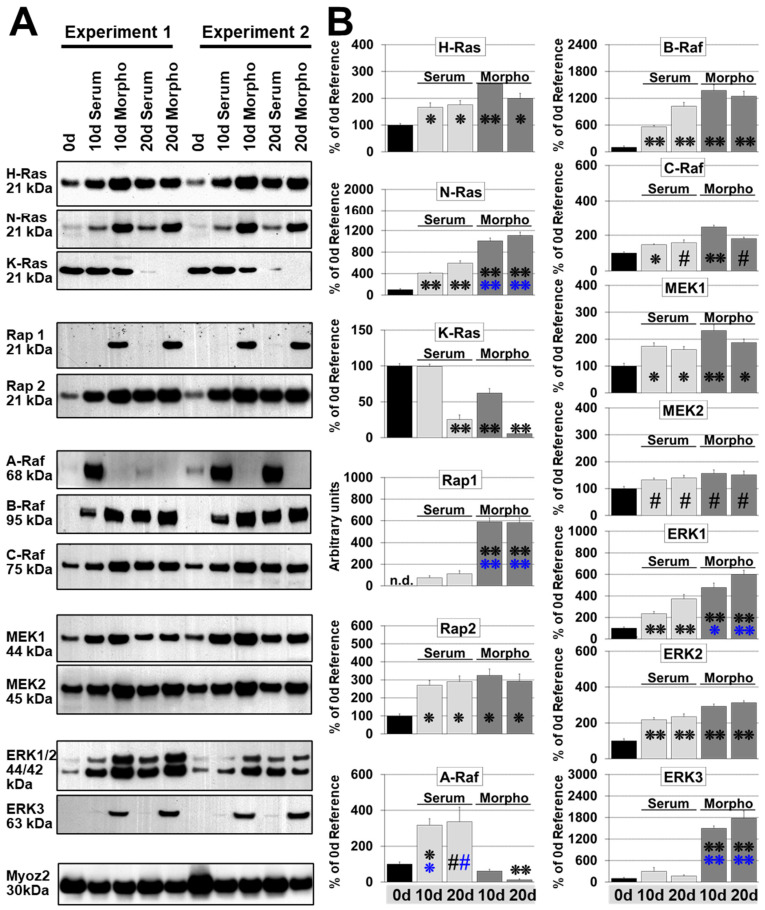
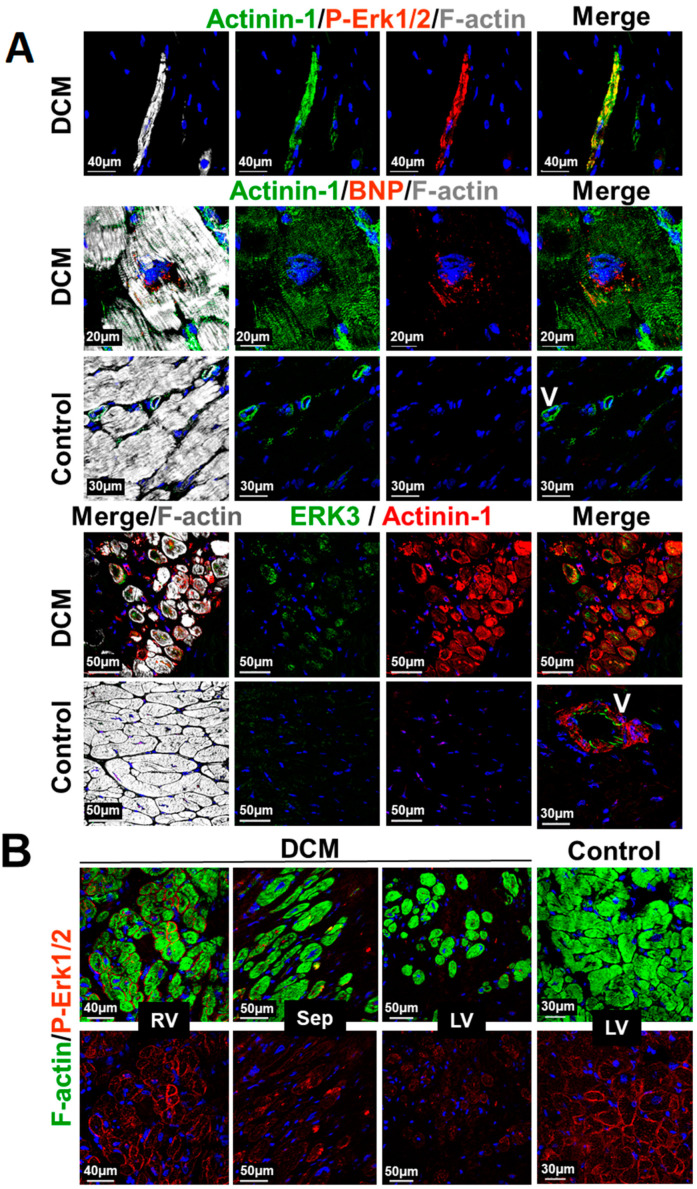
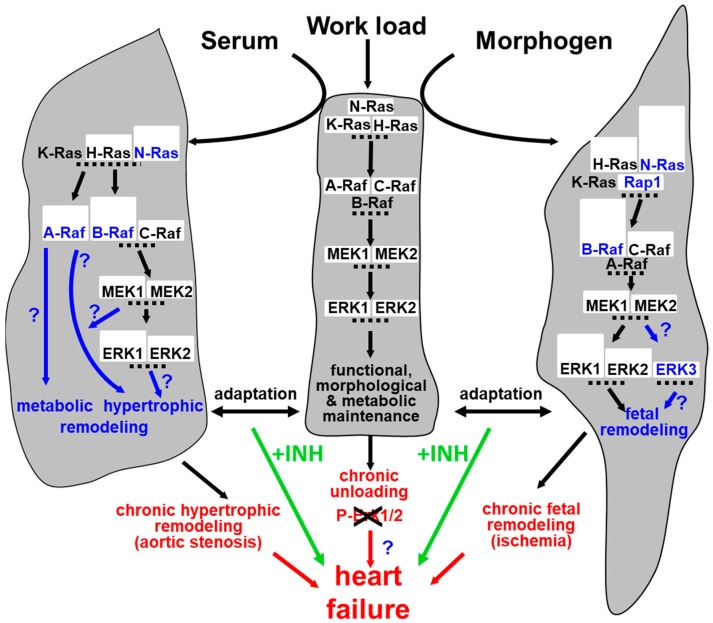
Fetal and hypertrophic remodeling are hallmarks of cardiac restructuring leading chronically to heart failure. Since the Ras/Raf/MEK/ERK cascade (MAPK) is involved in the development of heart failure, we hypothesized, first, that fetal remodeling is different from hypertrophy and, second, that remodeling of the MAPK occurs. To test our hypothesis, we analyzed models of cultured adult rat cardiomyocytes as well as investigated myocytes in the failing human myocardium by western blot and confocal microscopy. Fetal remodeling was induced through endothelial morphogens and monitored by the reexpression of Acta2, Actn1, and Actb. Serum-induced hypertrophy was determined by increased surface size and protein content of cardiomyocytes. Serum and morphogens caused reprogramming of Ras/Raf/MEK/ERK. In both models H-Ras, N-Ras, Rap2, B- and C-Raf, MEK1/2 as well as ERK1/2 increased while K-Ras was downregulated. Atrophy, MAPK-dependent ischemic resistance, loss of A-Raf, and reexpression of Rap1 and Erk3 highlighted fetal remodeling, while A-Raf accumulation marked hypertrophy. The knock-down of B-Raf by siRNA reduced MAPK activation and fetal reprogramming. In conclusion, we demonstrate that fetal and hypertrophic remodeling are independent processes and involve reprogramming of the MAPK.
Keywords: ERK; MEK; Raf; Rap; Ras; dedifferentiation; heart failure; hypertrophy; remodeling; reprogramming.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- Hein S., Arnon E., Kostin S., Schonburg M., Elsasser A., Polyakova V., Bauer E.P., Klovekorn W.-P., Schaper J. Progression From Compensated Hypertrophy to Failure in the Pressure-Overloaded Human Heart: Structural Deterioration and Compensatory Mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.CIR.0000051865.66123.B7. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
