

A defined structural unit enables de novo design of small-molecule-binding proteins
- PMID: 32883865
- PMCID: PMC7526616
- DOI: 10.1126/science.abb8330
A defined structural unit enables de novo design of small-molecule-binding proteins
Abstract
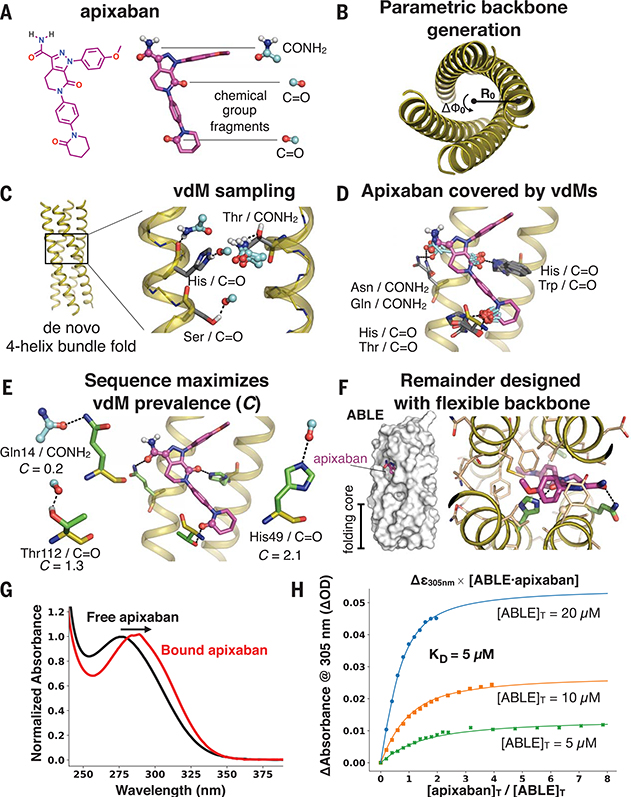
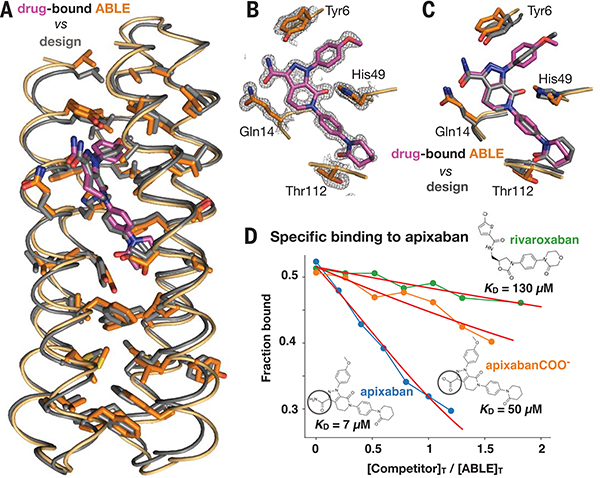
The de novo design of proteins that bind highly functionalized small molecules represents a great challenge. To enable computational design of binders, we developed a unit of protein structure-a van der Mer (vdM)-that maps the backbone of each amino acid to statistically preferred positions of interacting chemical groups. Using vdMs, we designed six de novo proteins to bind the drug apixaban; two bound with low and submicromolar affinity. X-ray crystallography and mutagenesis confirmed a structure with a precisely designed cavity that forms favorable interactions in the drug-protein complex. vdMs may enable design of functional proteins for applications in sensing, medicine, and catalysis.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
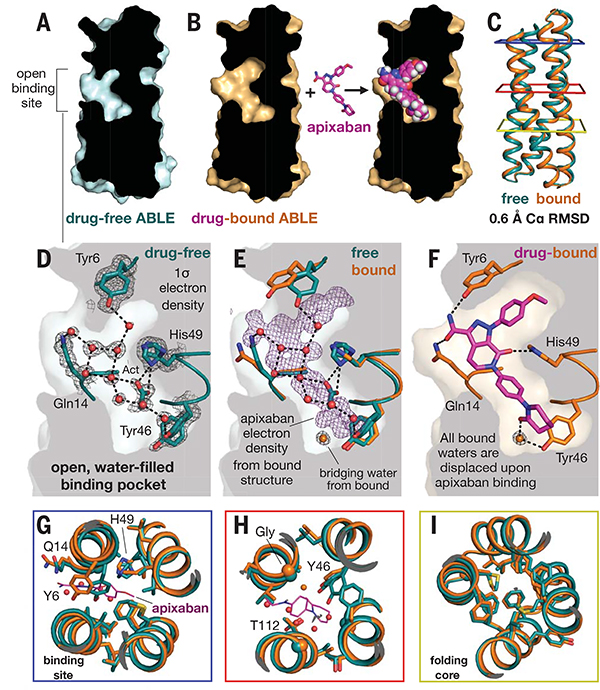
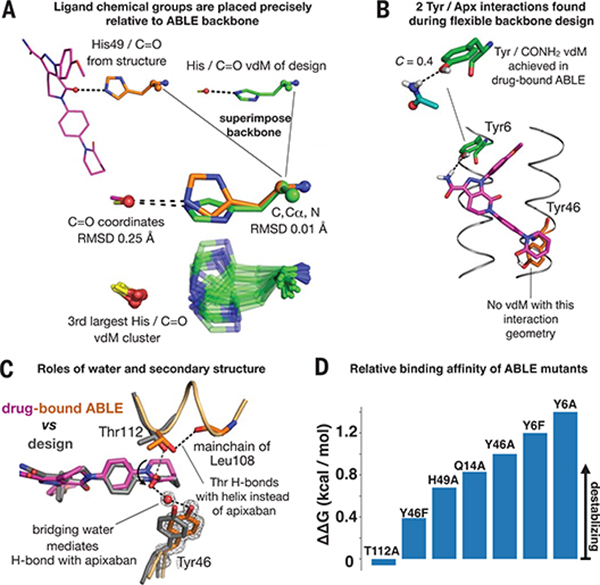
Comment in
-
Can proteins be truly designed sans function?Science. 2020 Sep 4;369(6508):1166-1167. doi: 10.1126/science.abd4791. Science. 2020. PMID: 32883850 No abstract available.
-
De novo design of small-molecule-binding proteins.Nat Methods. 2020 Nov;17(11):1073. doi: 10.1038/s41592-020-00995-3. Nat Methods. 2020. PMID: 33122857 No abstract available.
References
-
- Anfinsen CB, Science 181, 223–230 (1973). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
