

Patterns of Reliability: Assessing the Reproducibility and Integrity of DNA Methylation Measurement
- PMID: 32885222
- PMCID: PMC7467214
- DOI: 10.1016/j.patter.2020.100014
Patterns of Reliability: Assessing the Reproducibility and Integrity of DNA Methylation Measurement
Abstract
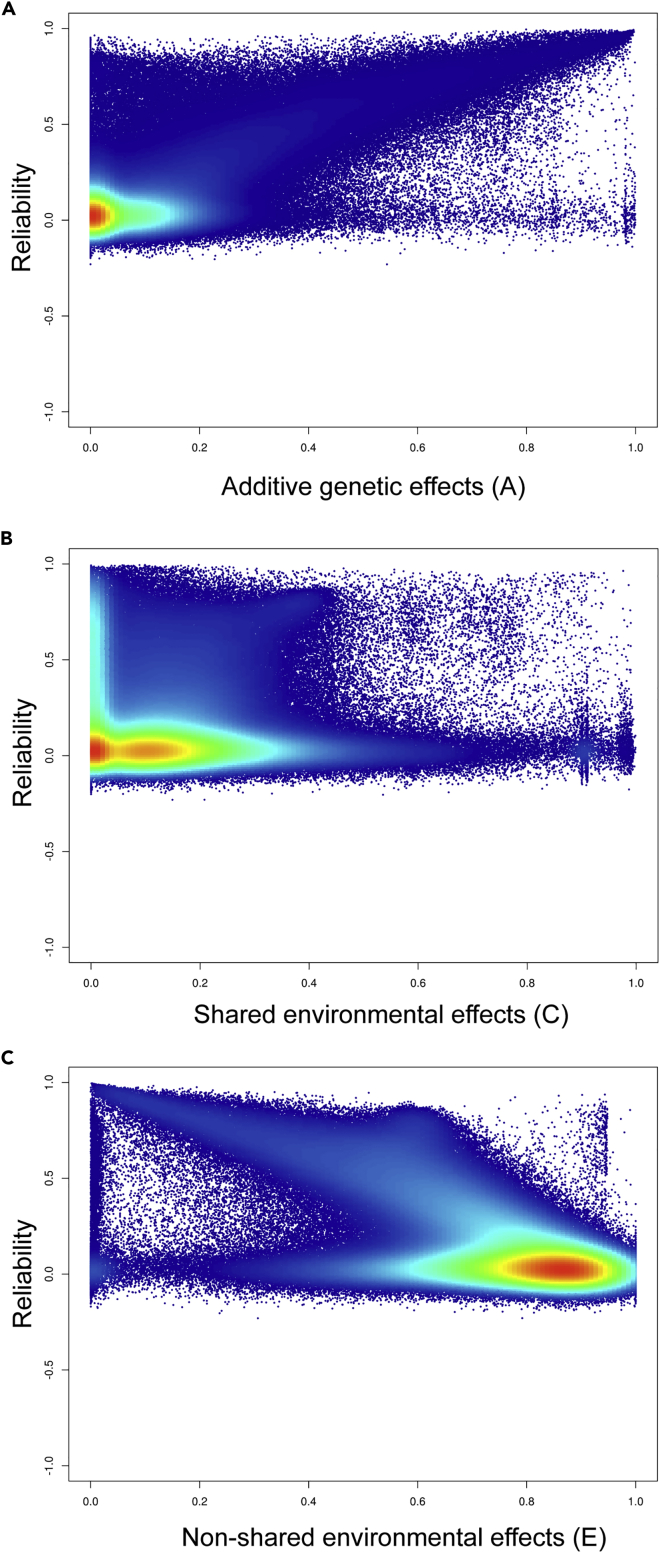
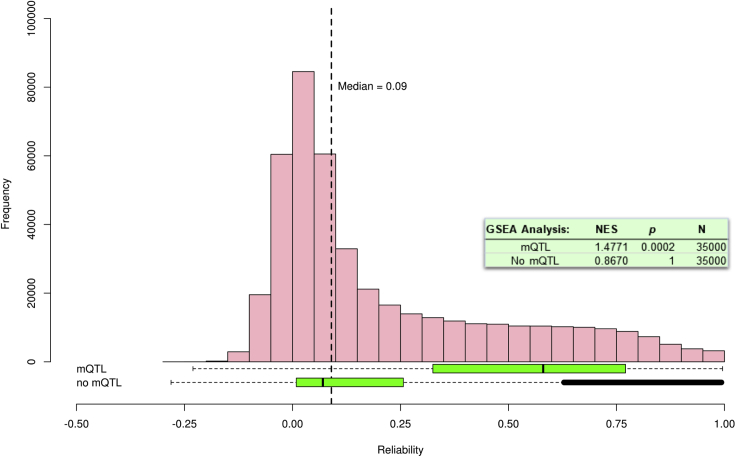
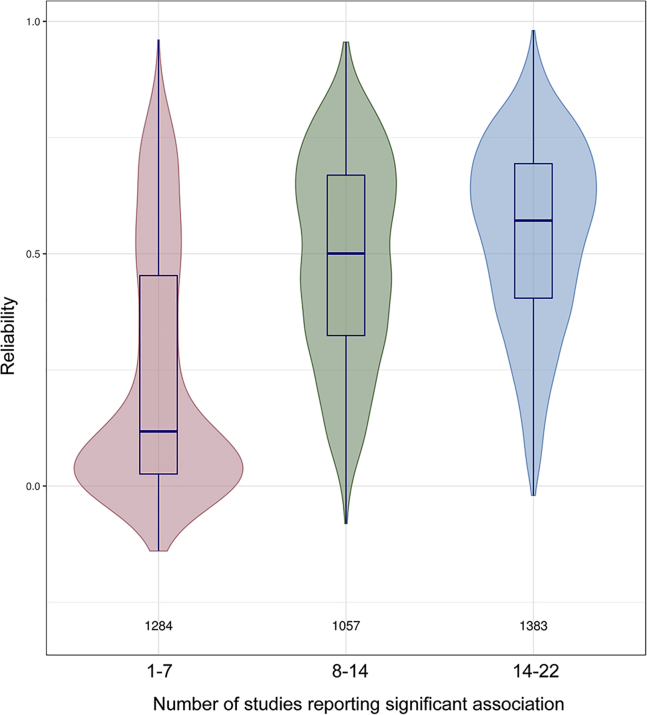
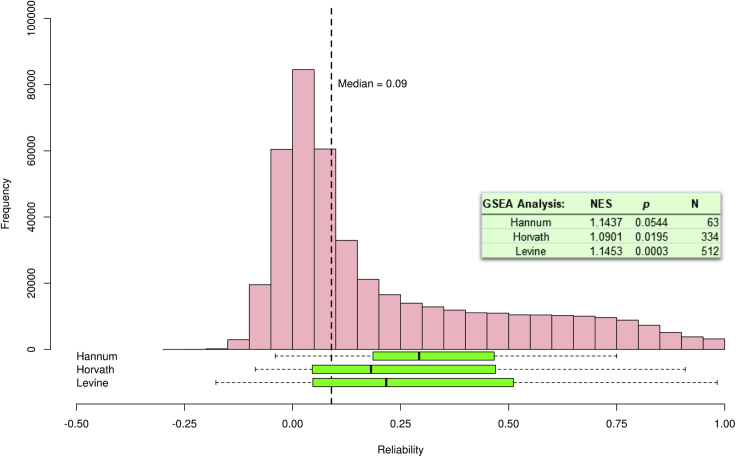
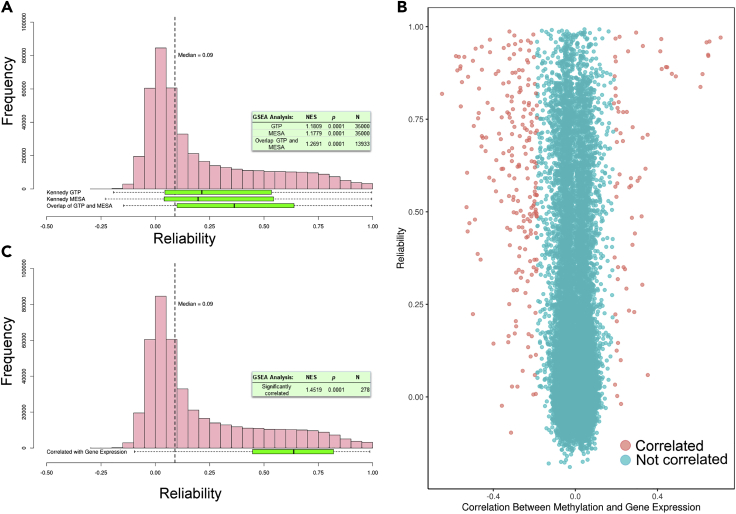
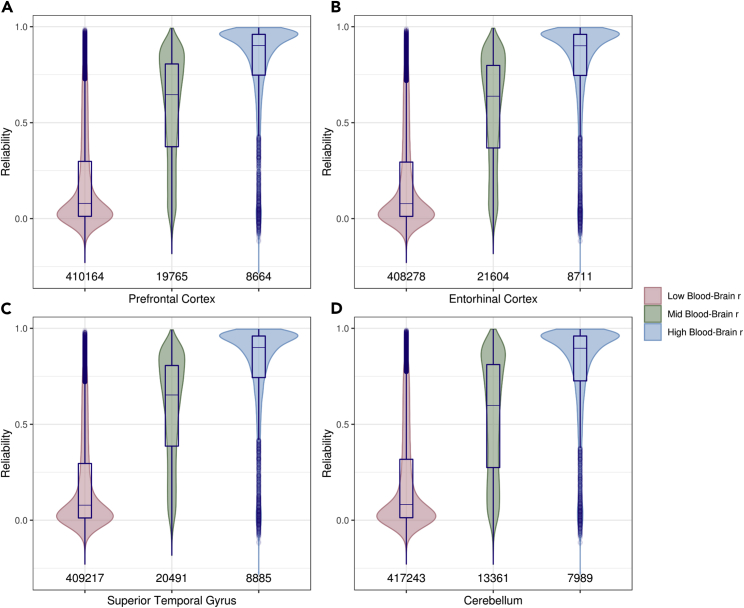
DNA methylation plays an important role in both normal human development and risk of disease. The most utilized method of assessing DNA methylation uses BeadChips, generating an epigenome-wide "snapshot" of >450,000 observations (probe measurements) per assay. However, the reliability of each of these measurements is not equal, and little consideration is paid to consequences for research. We correlated repeat measurements of the same DNA samples using the Illumina HumanMethylation450K and the Infinium MethylationEPIC BeadChips in 350 blood DNA samples. Probes that were reliably measured were more heritable and showed consistent associations with environmental exposures, gene expression, and greater cross-tissue concordance. Unreliable probes were less replicable and generated an unknown volume of false negatives. This serves as a lesson for working with DNA methylation data, but the lessons are equally applicable to working with other data: as we advance toward generating increasingly greater volumes of data, failure to document reliability risks harming reproducibility.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
-
- Robertson K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005;6:597–610. - PubMed
-
- Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. - PubMed
-
- Velasco G., Francastel C. Genetics meets DNA methylation in rare diseases. Clin. Genet. 2019;95:210–220. - PubMed
-
- Ruskin H.J., Barat A. Recent advances in computational epigenetics. Adv. Genomics Genet. 2018;8:12.
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
