

CRISPR-GEMM Pooled Mutagenic Screening Identifies KMT2D as a Major Modulator of Immune Checkpoint Blockade
- PMID: 32887696
- PMCID: PMC7710536
- DOI: 10.1158/2159-8290.CD-19-1448
CRISPR-GEMM Pooled Mutagenic Screening Identifies KMT2D as a Major Modulator of Immune Checkpoint Blockade
Abstract
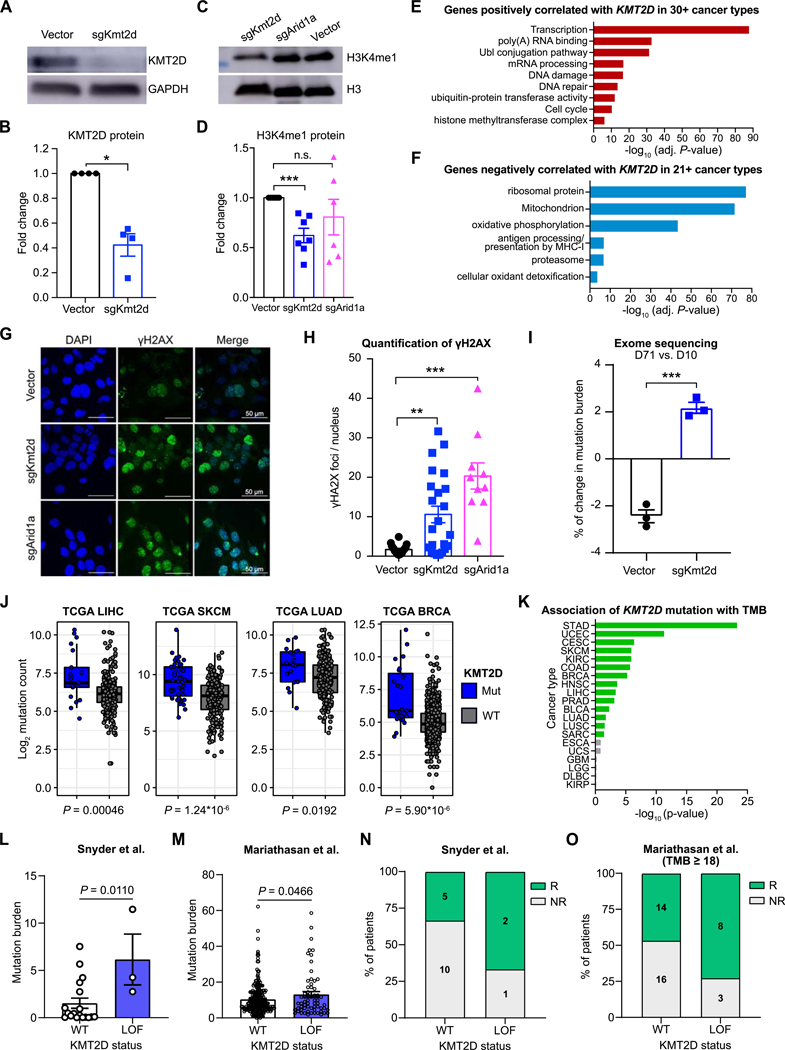
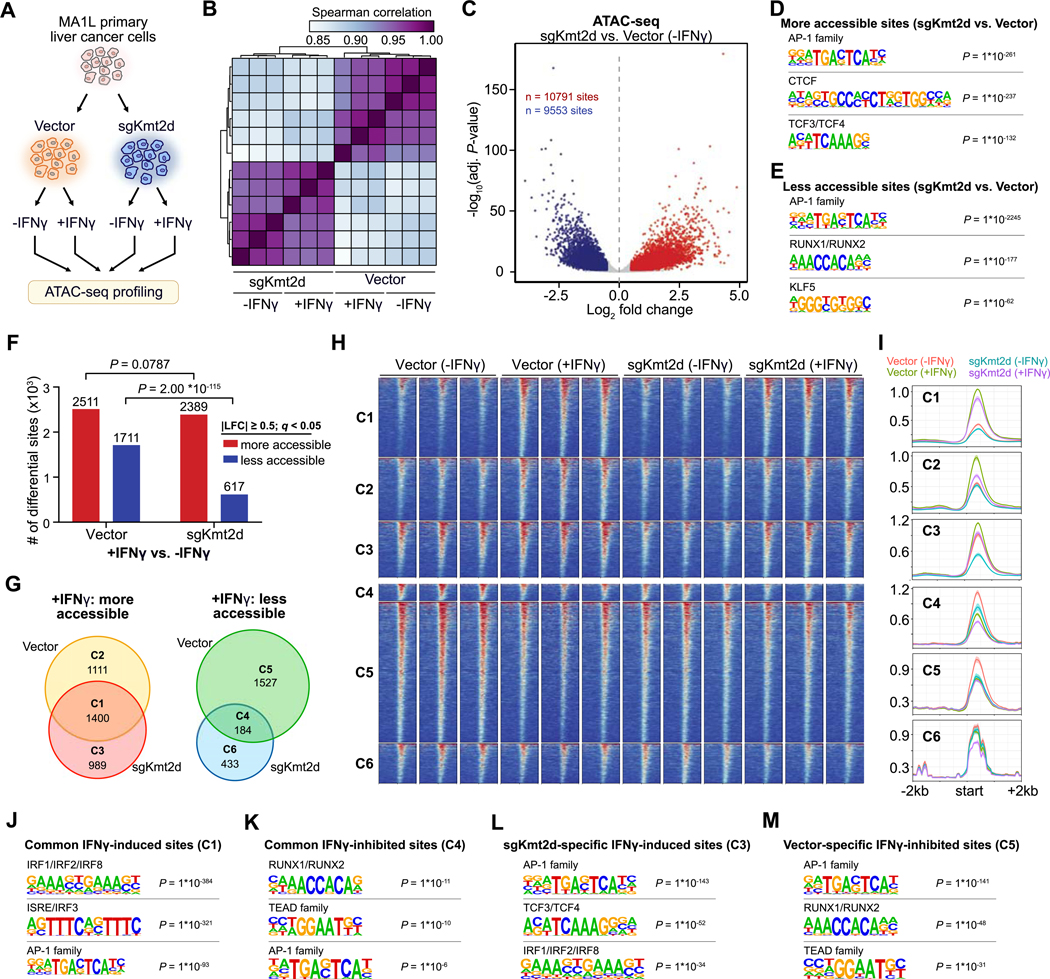
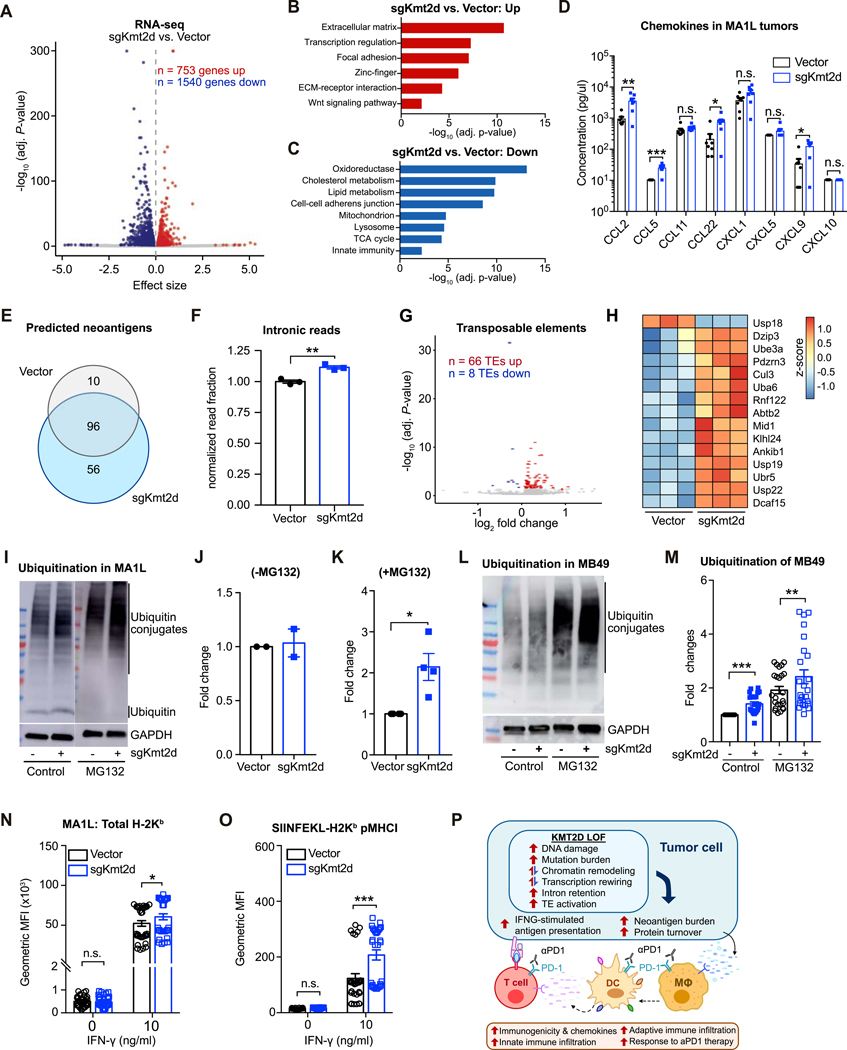
Immune checkpoint blockade (ICB) has shown remarkable clinical efficacy in several cancer types. However, only a fraction of patients will respond to ICB. Here, we performed pooled mutagenic screening with CRISPR-mediated genetically engineered mouse models (CRISPR-GEMM) in ICB settings, and identified KMT2D as a major modulator of ICB response across multiple cancer types. KMT2D encodes a histone H3K4 methyltransferase and is among the most frequently mutated genes in patients with cancer. Kmt2d loss led to increased DNA damage and mutation burden, chromatin remodeling, intron retention, and activation of transposable elements. In addition, Kmt2d-mutant cells exhibited increased protein turnover and IFNγ-stimulated antigen presentation. In turn, Kmt2d-mutant tumors in both mouse and human were characterized by increased immune infiltration. These data demonstrate that Kmt2d deficiency sensitizes tumors to ICB by augmenting tumor immunogenicity, and also highlight the power of CRISPR-GEMMs for interrogating complex molecular landscapes in immunotherapeutic contexts that preserve the native tumor microenvironment. SIGNIFICANCE: ICB is ineffective in the majority of patients. Through direct in vivo CRISPR mutagenesis screening in GEMMs of cancer, we find Kmt2d deficiency sensitizes tumors to ICB. Considering the prevalence of KMT2D mutations, this finding potentially has broad implications for patient stratification and clinical decision-making.This article is highlighted in the In This Issue feature, p. 1775.
©2020 American Association for Cancer Research.
Conflict of interest statement
Conflict of interest disclosure statement
The authors declare no competing interest related to this work. SC is a co-founder, funding recipient and scientific advisor of EvolveImmune Therapeutics, which is not related to this study.
Figures
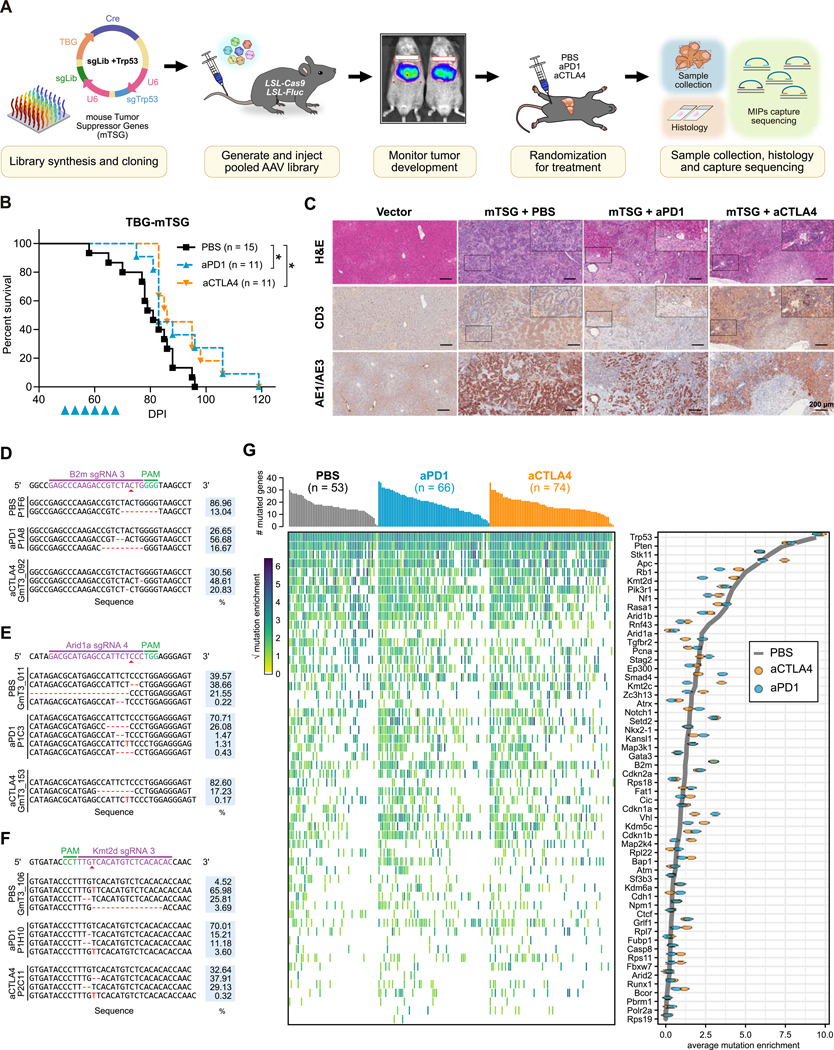
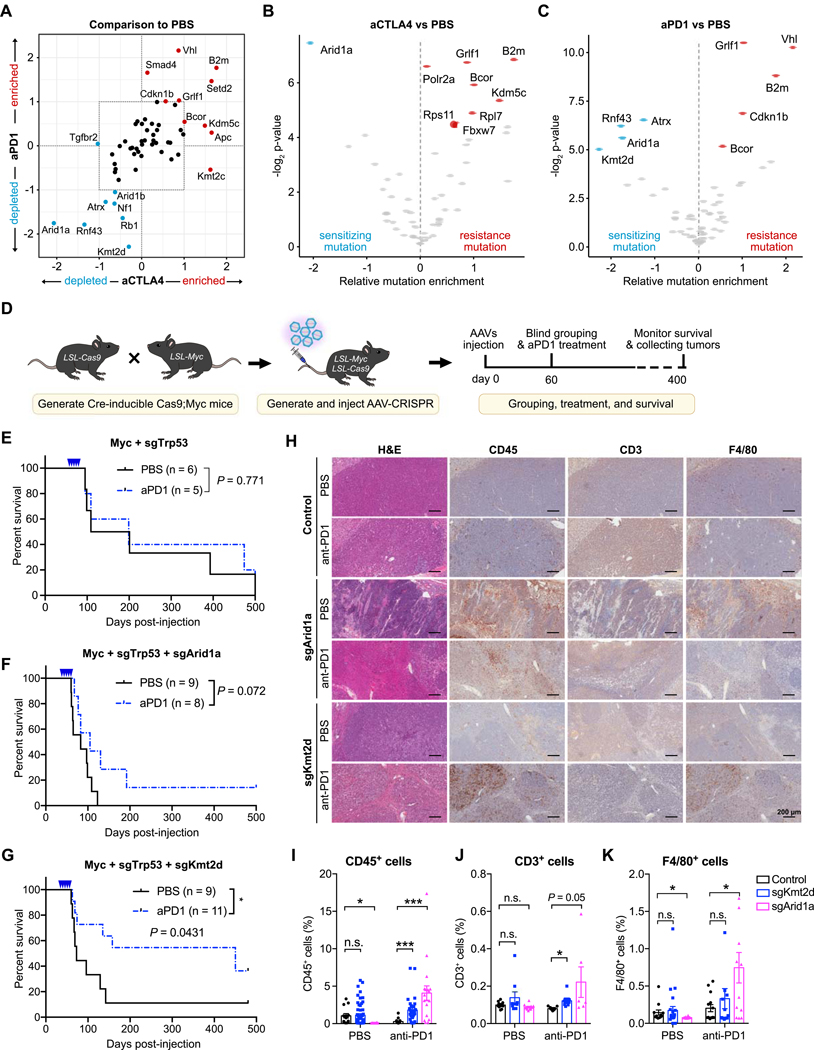
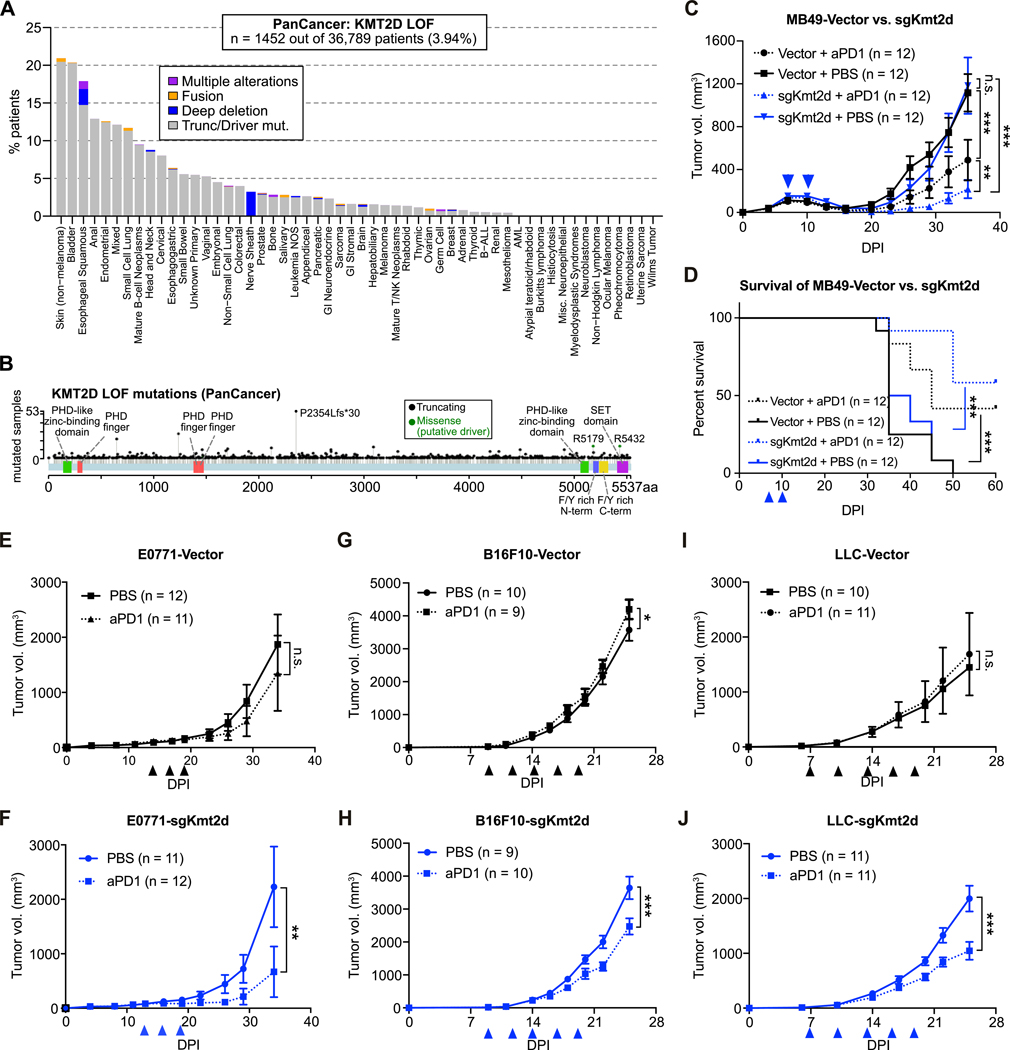
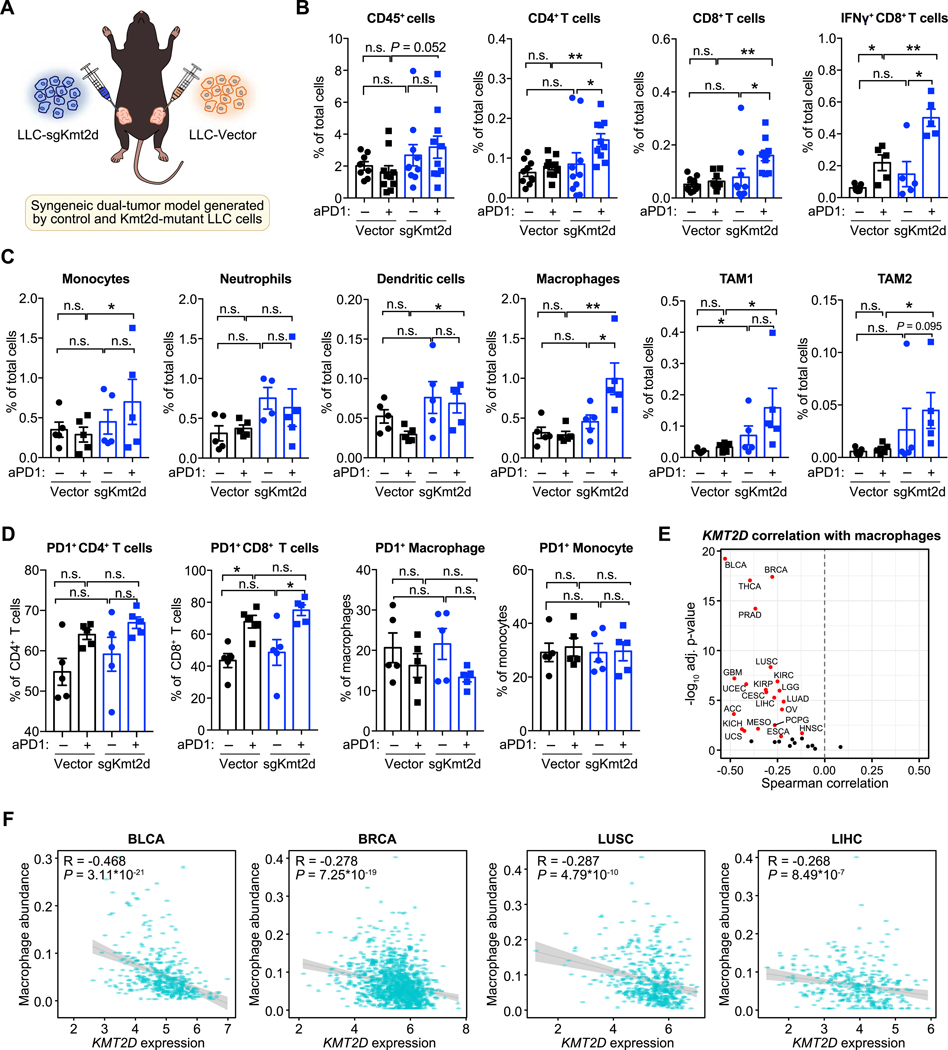
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
