

Impact of DNA extraction on whole genome sequencing analysis for characterization and relatedness of Shiga toxin-producing Escherichia coli isolates
- PMID: 32887913
- PMCID: PMC7474065
- DOI: 10.1038/s41598-020-71207-3
Impact of DNA extraction on whole genome sequencing analysis for characterization and relatedness of Shiga toxin-producing Escherichia coli isolates
Abstract
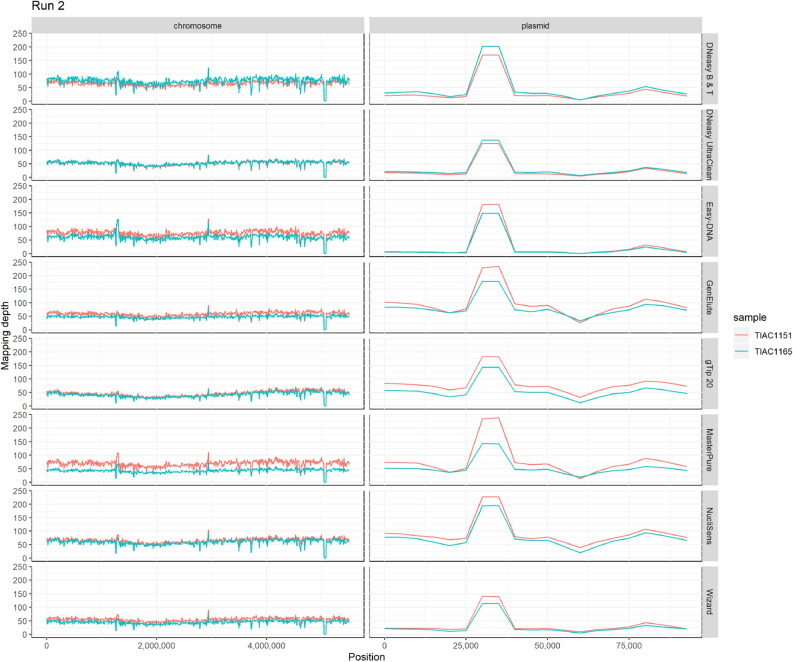
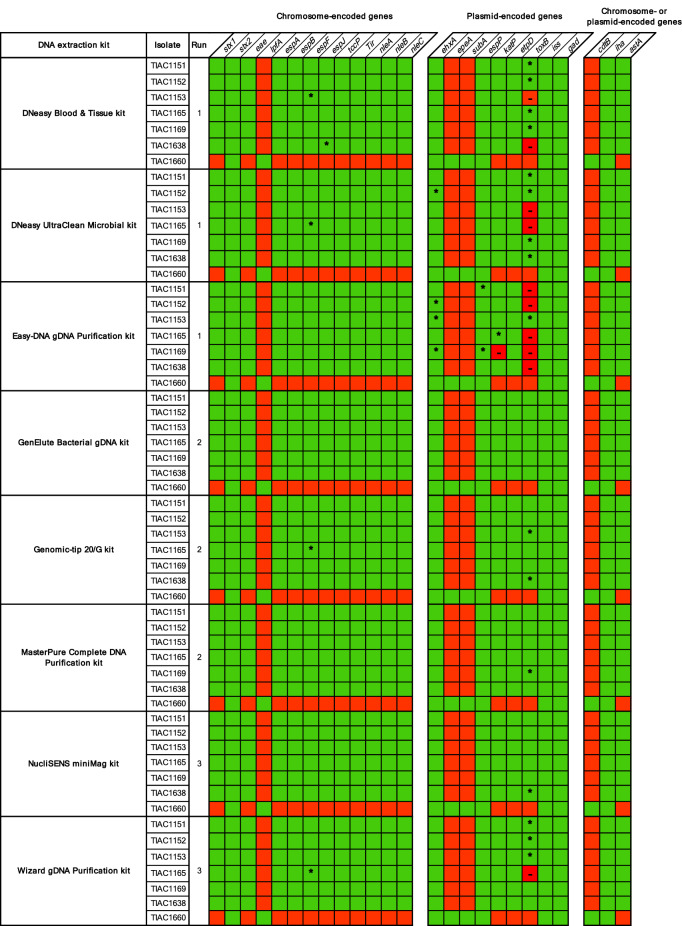
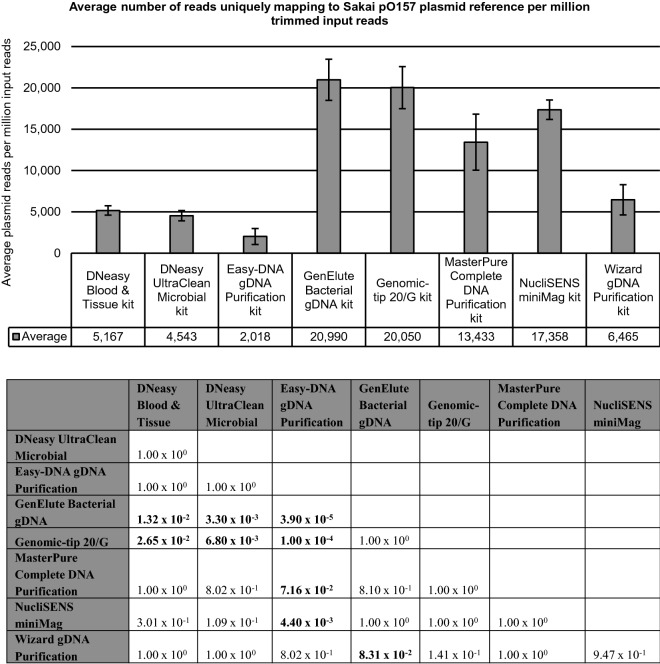
Whole genome sequencing (WGS) has proven to be the ultimate tool for bacterial isolate characterization and relatedness determination. However, standardized and harmonized workflows, e.g. for DNA extraction, are required to ensure robust and exchangeable WGS data. Data sharing between (inter)national laboratories is essential to support foodborne pathogen control, including outbreak investigation. This study evaluated eight commercial DNA preparation kits for their potential influence on: (i) DNA quality for Nextera XT library preparation; (ii) MiSeq sequencing (data quality, read mapping against plasmid and chromosome references); and (iii) WGS data analysis, i.e. isolate characterization (serotyping, virulence and antimicrobial resistance genotyping) and phylogenetic relatedness (core genome multilocus sequence typing and single nucleotide polymorphism analysis). Shiga toxin-producing Escherichia coli (STEC) was selected as a case study. Overall, data quality and inferred phylogenetic relationships between isolates were not affected by the DNA extraction kit choice, irrespective of the presence of confounding factors such as EDTA in DNA solution buffers. Nevertheless, completeness of STEC characterization was, although not substantially, influenced by the plasmid extraction performance of the kits, especially when using Nextera XT library preparation. This study contributes to addressing the WGS challenges of standardizing protocols to support data portability and to enable full exploitation of its potential.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- García Fierro, R. et al. Outcome of EC/EFSA questionnaire (2016) on use of whole genome sequencing (WGS) for food- and waterborne pathogens isolated from animals, food, feed and related environmental samples in EU/EFTA countries. EFSA J.15, 2018. 10.2903/sp.efsa.2018.EN-1432 (2016).
-
- ECDC. Monitoring the Use of Whole-Genome Sequencing in Infectious Disease Surveillance in Europe. (2018). 10.2900/037665.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
