

Mutations of the Transcriptional Corepressor ZMYM2 Cause Syndromic Urinary Tract Malformations
- PMID: 32891193
- PMCID: PMC7536580
- DOI: 10.1016/j.ajhg.2020.08.013
Mutations of the Transcriptional Corepressor ZMYM2 Cause Syndromic Urinary Tract Malformations
Abstract
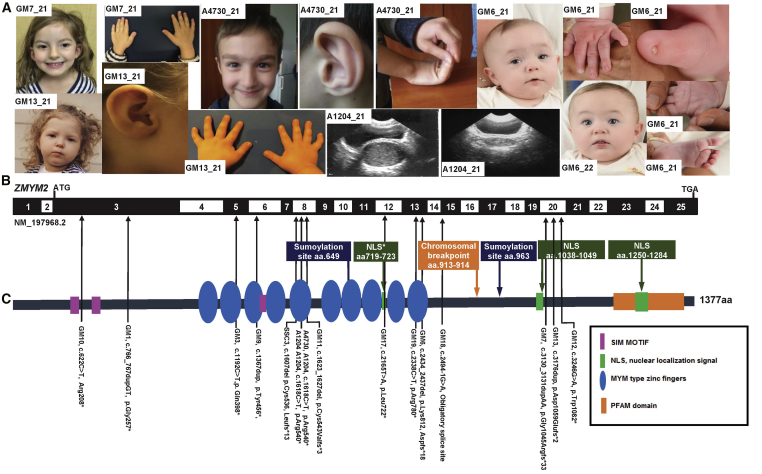
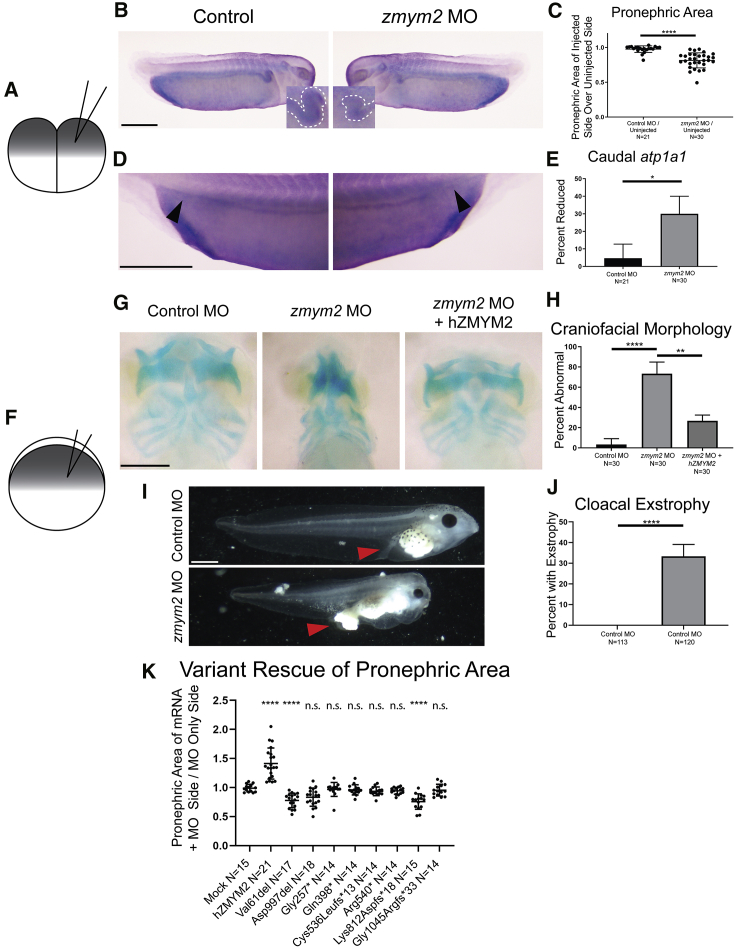
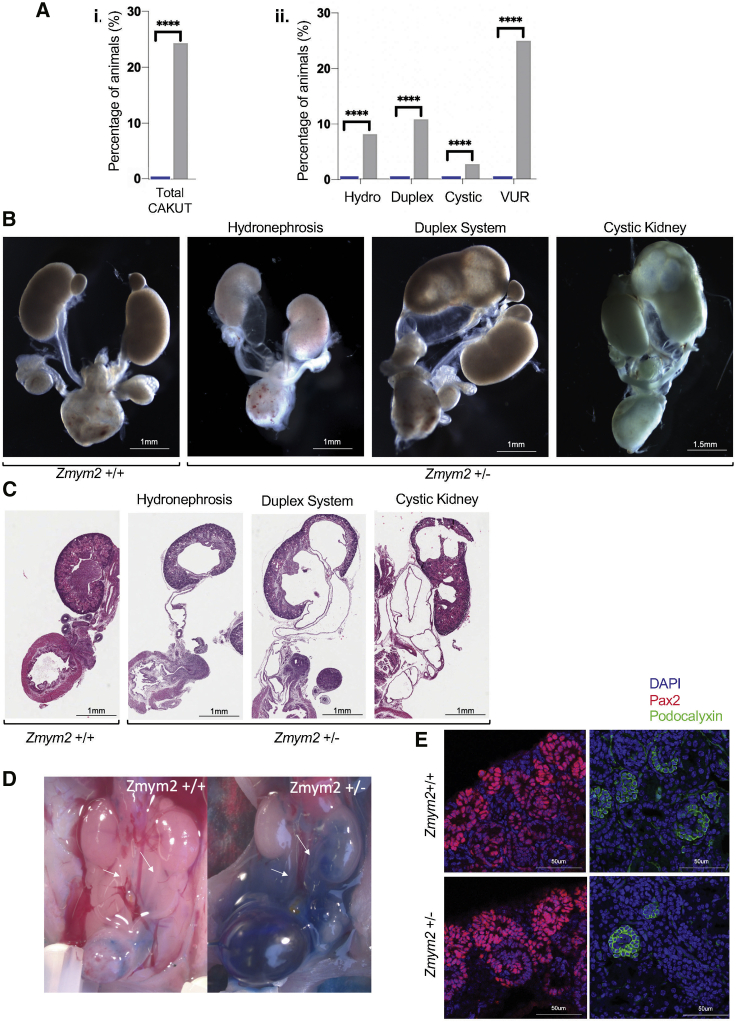
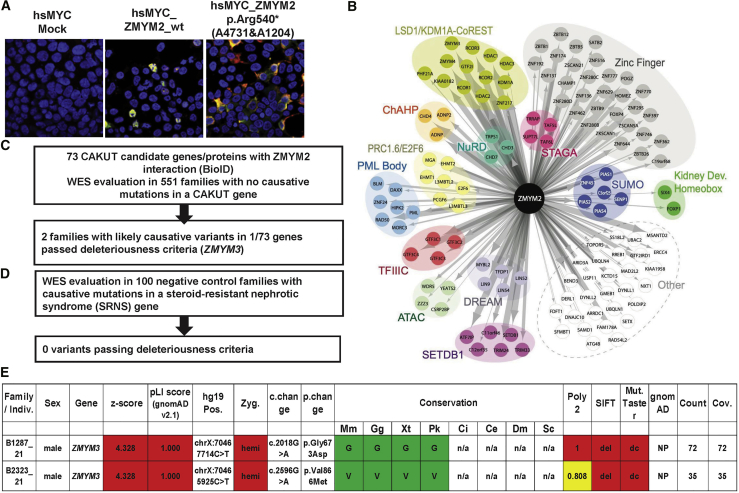
Congenital anomalies of the kidney and urinary tract (CAKUT) constitute one of the most frequent birth defects and represent the most common cause of chronic kidney disease in the first three decades of life. Despite the discovery of dozens of monogenic causes of CAKUT, most pathogenic pathways remain elusive. We performed whole-exome sequencing (WES) in 551 individuals with CAKUT and identified a heterozygous de novo stop-gain variant in ZMYM2 in two different families with CAKUT. Through collaboration, we identified in total 14 different heterozygous loss-of-function mutations in ZMYM2 in 15 unrelated families. Most mutations occurred de novo, indicating possible interference with reproductive function. Human disease features are replicated in X. tropicalis larvae with morpholino knockdowns, in which expression of truncated ZMYM2 proteins, based on individual mutations, failed to rescue renal and craniofacial defects. Moreover, heterozygous Zmym2-deficient mice recapitulated features of CAKUT with high penetrance. The ZMYM2 protein is a component of a transcriptional corepressor complex recently linked to the silencing of developmentally regulated endogenous retrovirus elements. Using protein-protein interaction assays, we show that ZMYM2 interacts with additional epigenetic silencing complexes, as well as confirming that it binds to FOXP1, a transcription factor that has also been linked to CAKUT. In summary, our findings establish that loss-of-function mutations of ZMYM2, and potentially that of other proteins in its interactome, as causes of human CAKUT, offering new routes for studying the pathogenesis of the disorder.
Keywords: FIM; ZMYM2; ZNF198; congenital anomalies of the kidney and urinary tract; extra-renal features; genetic kidney disease; genomic analysis; syndromic CAKUT; transcription regulator; whole-exome sequencing.
Copyright © 2020 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- van der Ven A.T., Connaughton D.M., Ityel H., Mann N., Nakayama M., Chen J., Vivante A., Hwang D.Y., Schulz J., Braun D.A. Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract. J. Am. Soc. Nephrol. 2018;29:2348–2361. - PMC - PubMed
-
- Weber S., Moriniere V., Knüppel T., Charbit M., Dusek J., Ghiggeri G.M., Jankauskiené A., Mir S., Montini G., Peco-Antic A. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J. Am. Soc. Nephrol. 2006;17:2864–2870. - PubMed
-
- Rasmussen M., Sunde L., Nielsen M.L., Ramsing M., Petersen A., Hjortshøj T.D., Olsen T.E., Tabor A., Hertz J.M., Johnsen I. Targeted gene sequencing and whole-exome sequencing in autopsied fetuses with prenatally diagnosed kidney anomalies. Clin. Genet. 2018;93:860–869. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- MC_UU_00007/3/MRC_/Medical Research Council/United Kingdom
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- P20 DK116191/DK/NIDDK NIH HHS/United States
- P30 DK079310/DK/NIDDK NIH HHS/United States
- R01 DK078226/DK/NIDDK NIH HHS/United States
- T32 GM007205/GM/NIGMS NIH HHS/United States
- R01 HD102186/HD/NICHD NIH HHS/United States
- R01 DK103184/DK/NIDDK NIH HHS/United States
- R01 DK115574/DK/NIDDK NIH HHS/United States
- R01 DK088767/DK/NIDDK NIH HHS/United States
- R01 DK133940/DK/NIDDK NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- P50 HD103524/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
