

Metabolic and functional specialisations of the pancreatic beta cell: gene disallowance, mitochondrial metabolism and intercellular connectivity
- PMID: 32894309
- PMCID: PMC7476987
- DOI: 10.1007/s00125-020-05205-5
Metabolic and functional specialisations of the pancreatic beta cell: gene disallowance, mitochondrial metabolism and intercellular connectivity
Abstract
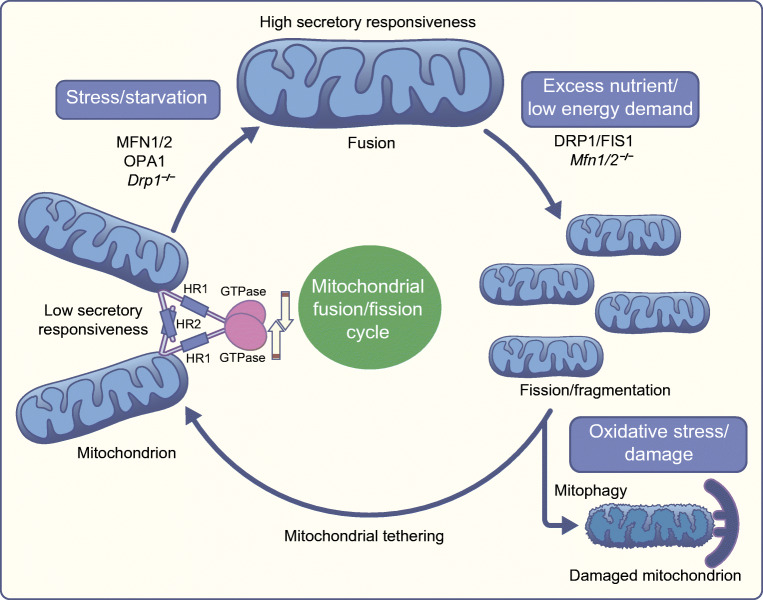
All forms of diabetes mellitus involve the loss or dysfunction of pancreatic beta cells, with the former predominating in type 1 diabetes and the latter in type 2 diabetes. Deeper understanding of the coupling mechanisms that link glucose metabolism in these cells to the control of insulin secretion is therefore likely to be essential to develop new therapies. Beta cells display a remarkable metabolic specialisation, expressing high levels of metabolic sensing enzymes, including the glucose transporter GLUT2 (encoded by SLC2A2) and glucokinase (encoded by GCK). Genetic evidence flowing from both monogenic forms of diabetes and genome-wide association studies for the more common type 2 diabetes, supports the importance for normal glucose-stimulated insulin secretion of metabolic signalling via altered ATP generation, while also highlighting unsuspected roles for Zn2+ storage, intracellular lipid transfer and other processes. Intriguingly, genes involved in non-oxidative metabolic fates of the sugar, such as those for lactate dehydrogenase (LDHA) and monocarboxylate transporter-1 ([MCT-1] SLC16A1), as well as the acyl-CoA thioesterase (ACOT7) and others, are selectively repressed ('disallowed') in beta cells. Furthermore, mutations in genes critical for mitochondrial oxidative metabolism, such as TRL-CAG1-7 encoding tRNALeu, are linked to maternally inherited forms of diabetes. Correspondingly, impaired Ca2+ uptake into mitochondria, or collapse of a normally interconnected mitochondrial network, are associated with defective insulin secretion. Here, we suggest that altered mitochondrial metabolism may also impair beta cell-beta cell communication. Thus, we argue that defective oxidative glucose metabolism is central to beta cell failure in diabetes, acting both at the level of single beta cells and potentially across the whole islet to impair insulin secretion. Graphical abstract.
Keywords: Beta cells; Disallowed genes; Insulin secretion; Interconnectivity; Mitochondria; Review; Type 2 diabetes.
Figures




References
-
- McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Mol Genet Metab. 2011;104(4):648–653. doi: 10.1016/j.ymgme.2011.08.026. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
