

Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease
- PMID: 32897178
- PMCID: PMC7573242
- DOI: 10.1080/07391102.2020.1815584
Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease
Abstract
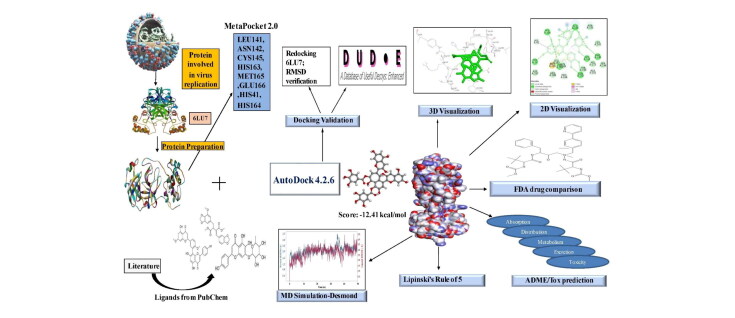
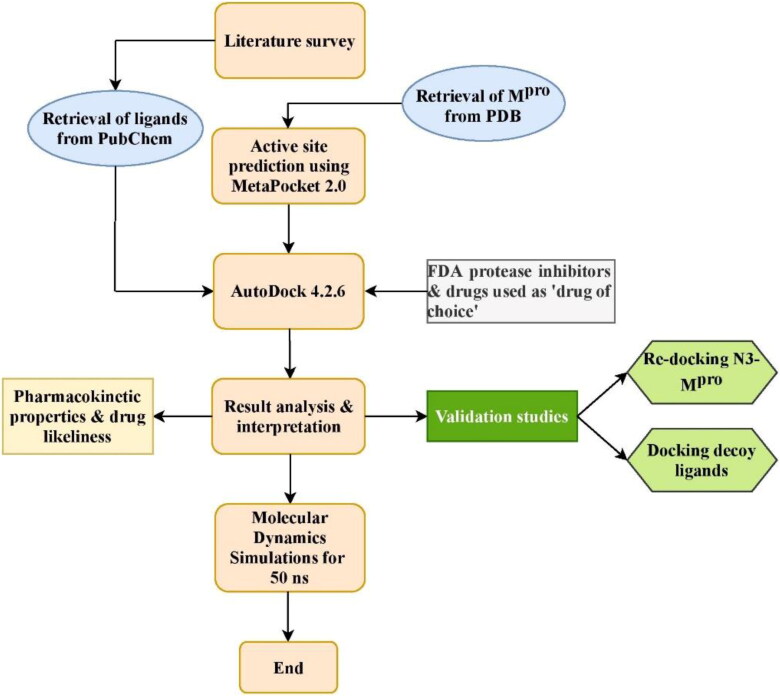
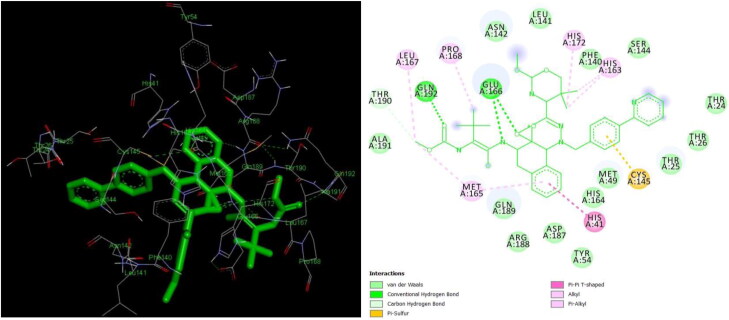
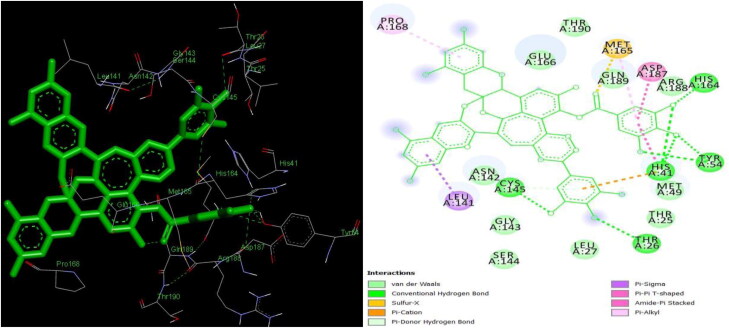
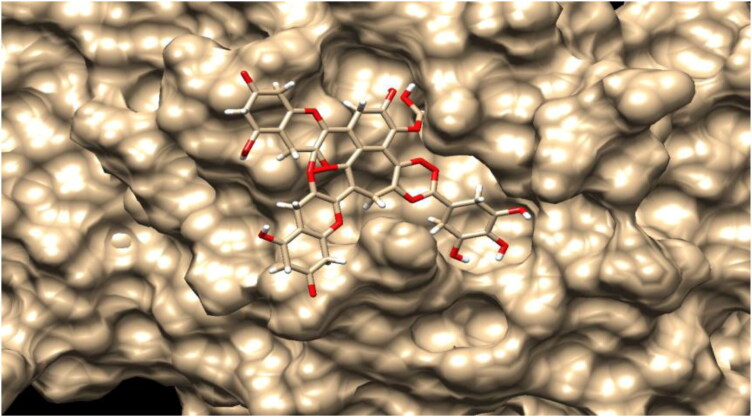
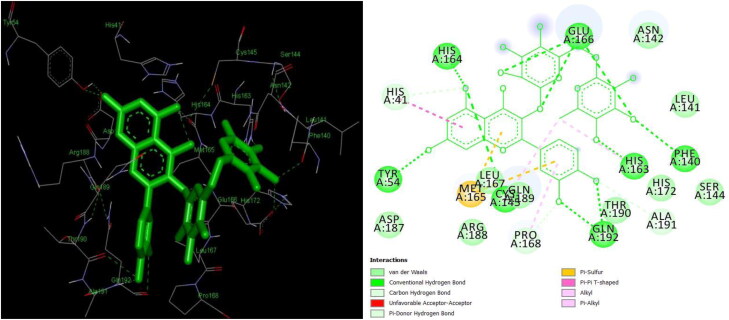
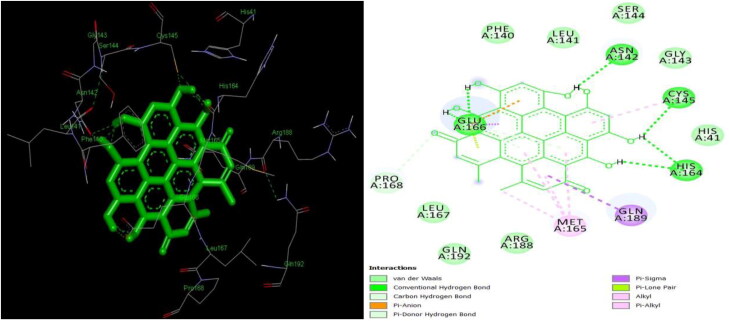
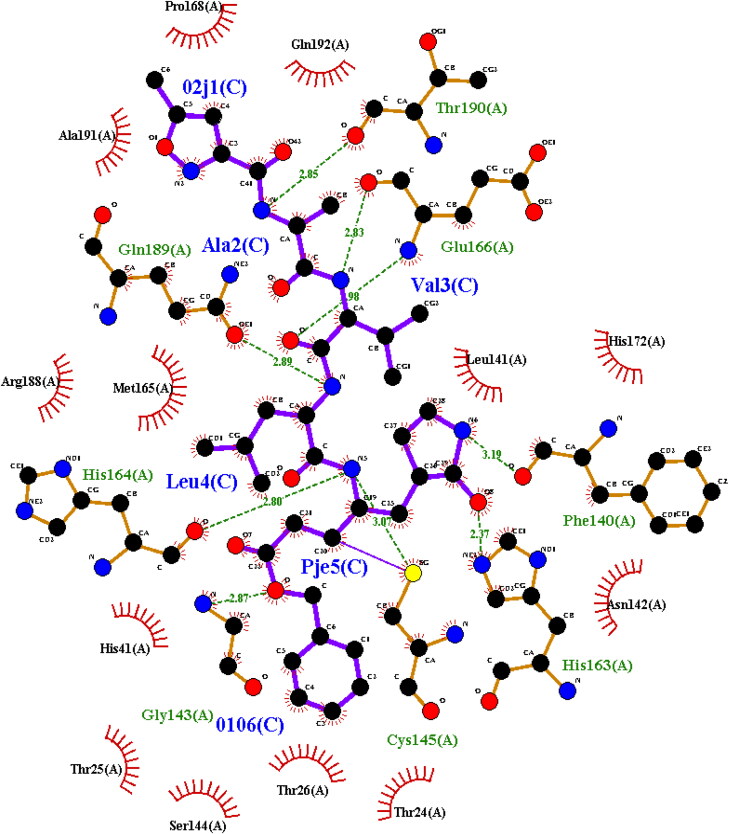
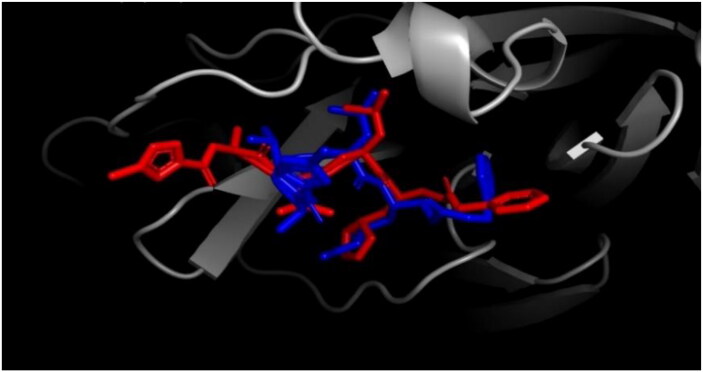
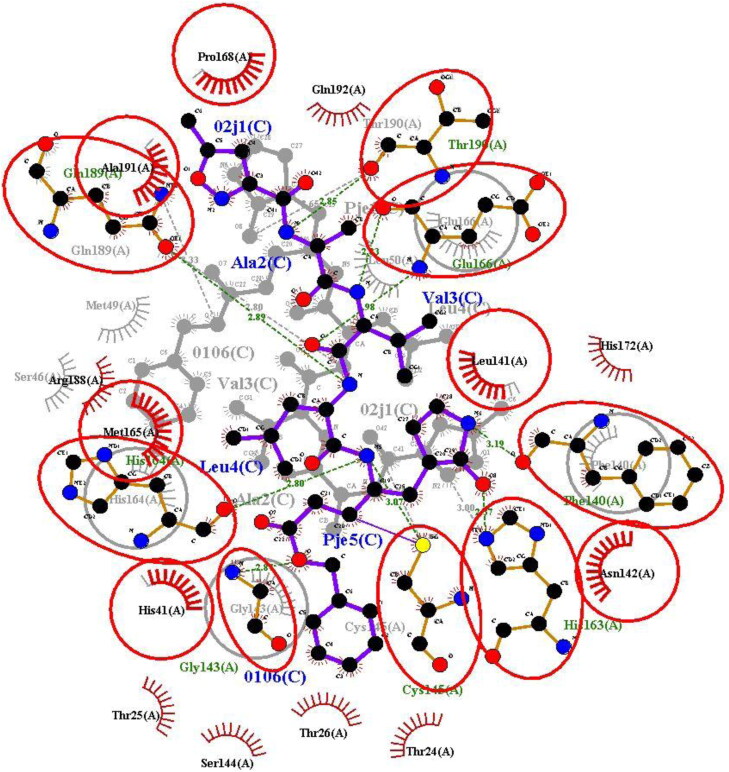
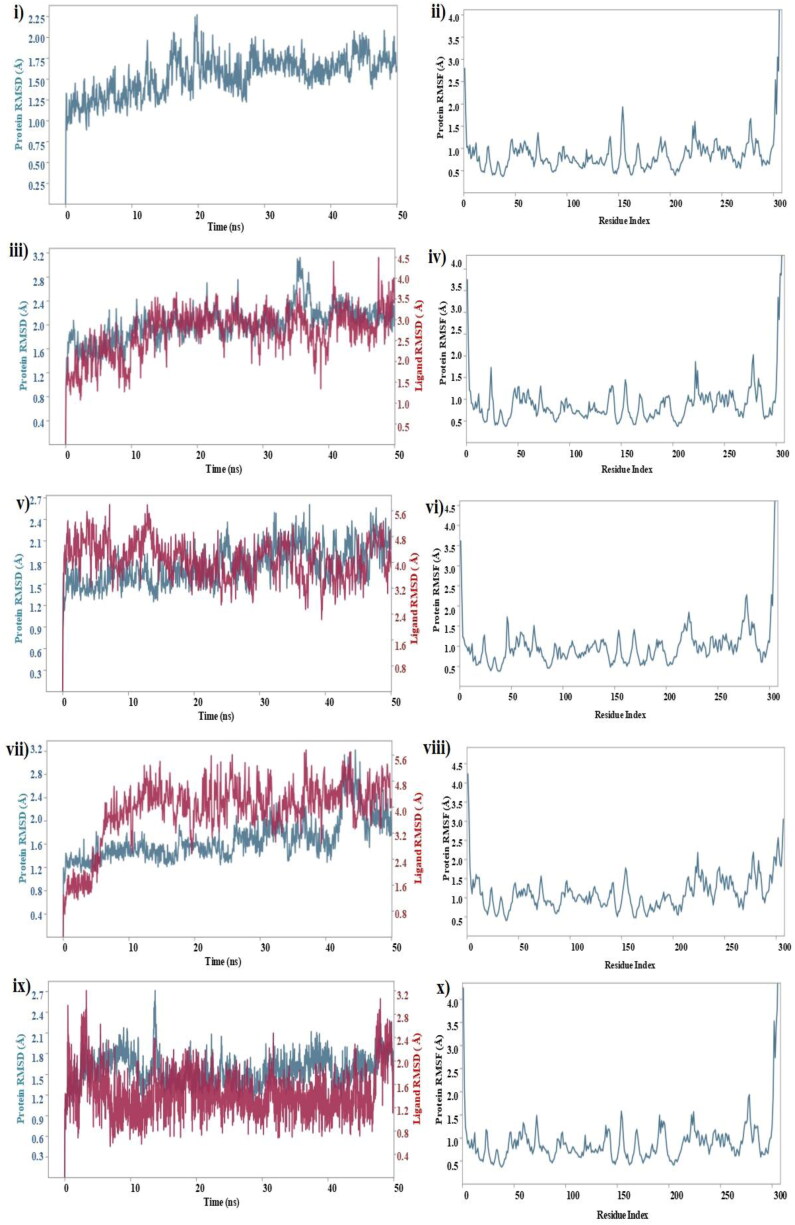
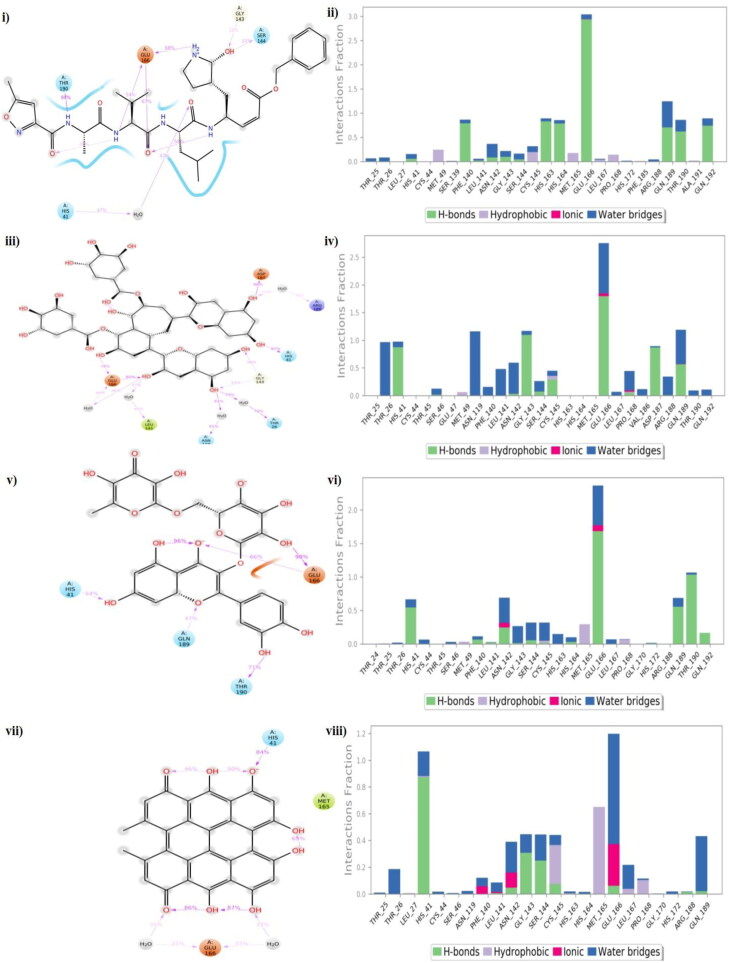
The study aims to evaluate the potency of two hundred natural antiviral phytocompounds against the active site of the Severe Acquired Respiratory Syndrome - Coronavirus - 2 (SARS-CoV-2) Main-Protease (Mpro) using AutoDock 4.2.6. The three- dimensional crystal structure of the Mpro (PDB Id: 6LU7) was retrieved from the Protein Data Bank (PDB), the active site was predicted using MetaPocket 2.0. Food and Drug Administration (FDA) approved viral protease inhibitors were used as standards for comparison of results. The compounds theaflavin-3-3'-digallate, rutin, hypericin, robustaflavone, and (-)-solenolide A with respective binding energy of -12.41 (Ki = 794.96 pM); -11.33 (Ki = 4.98 nM); -11.17 (Ki = 6.54 nM); -10.92 (Ki = 9.85 nM); and -10.82 kcal/mol (Ki = 11.88 nM) were ranked top as Coronavirus Disease - 2019 (COVID-19) Mpro inhibitors. The interacting amino acid residues were visualized using Discovery Studio 3.5 to elucidate the 2-dimensional and 3-dimensional interactions. The study was validated by i) re-docking the N3-peptide inhibitor-Mpro and superimposing them onto co-crystallized complex and ii) docking decoy ligands to Mpro. The ligands that showed low binding energy were further predicted for and pharmacokinetic properties and Lipinski's rule of 5 and the results are tabulated and discussed. Molecular dynamics simulations were performed for 50 ns for those compounds using the Desmond package, Schrödinger to assess the conformational stability and fluctuations of protein-ligand complexes during the simulation. Thus, the natural compounds could act as a lead for the COVID-19 regimen after in-vitro and in- vivo clinical trials.Communicated by Ramaswamy H. Sarma.
Keywords: Antiviral phytocompounds; COVID-19; SARS-CoV-2; decoy ligands; main-protease; molecular dynamics simulations; pharmacokinetic properties.
Conflict of interest statement
The authors declare a conflict of interest as none.
Figures
Similar articles
-
Virtual screening, ADMET prediction and dynamics simulation of potential compounds targeting the main protease of SARS-CoV-2.J Biomol Struct Dyn. 2021 Oct;39(17):6617-6632. doi: 10.1080/07391102.2020.1796812. Epub 2020 Jul 25. J Biomol Struct Dyn. 2021. PMID: 32715956 Free PMC article.
-
Protein reliability analysis and virtual screening of natural inhibitors for SARS-CoV-2 main protease (Mpro) through docking, molecular mechanic & dynamic, and ADMET profiling.J Biomol Struct Dyn. 2021 Oct;39(17):6810-6827. doi: 10.1080/07391102.2020.1806930. Epub 2020 Aug 14. J Biomol Struct Dyn. 2021. PMID: 32795148 Free PMC article.
-
In silico validation of coumarin derivatives as potential inhibitors against Main Protease, NSP10/NSP16-Methyltransferase, Phosphatase and Endoribonuclease of SARS CoV-2.J Biomol Struct Dyn. 2021 Nov;39(18):7306-7321. doi: 10.1080/07391102.2020.1808075. Epub 2020 Aug 24. J Biomol Struct Dyn. 2021. PMID: 32835632 Free PMC article.
-
Repurposing dye ligands as antivirals via a docking approach on viral membrane and globular proteins - SARS-CoV-2 and HPV-16.Biochim Biophys Acta Biomembr. 2024 Jan;1866(1):184220. doi: 10.1016/j.bbamem.2023.184220. Epub 2023 Aug 30. Biochim Biophys Acta Biomembr. 2024. PMID: 37657640 Review.
-
Methylxanthines as Potential Inhibitor of SARS-CoV-2: an In Silico Approach.Curr Pharmacol Rep. 2022;8(2):149-170. doi: 10.1007/s40495-021-00276-3. Epub 2022 Mar 8. Curr Pharmacol Rep. 2022. PMID: 35281252 Free PMC article. Review.
Cited by
-
Propolis, Bee Honey, and Their Components Protect against Coronavirus Disease 2019 (COVID-19): A Review of In Silico, In Vitro, and Clinical Studies.Molecules. 2021 Feb 25;26(5):1232. doi: 10.3390/molecules26051232. Molecules. 2021. PMID: 33669054 Free PMC article. Review.
-
Synthesis, crystal structure and in silico studies of novel 2,4-dimethoxy-tetrahydropyrimido[4,5-b]quinolin-6(7H)-ones.RSC Adv. 2022 Jun 29;12(29):18806-18820. doi: 10.1039/d2ra02694e. eCollection 2022 Jun 22. RSC Adv. 2022. PMID: 35873341 Free PMC article.
-
Docking Studies and Molecular Dynamics Simulations of Potential Inhibitors from the Brown Seaweed Sargassum polycystum (Phaeophyceae) against PLpro of SARS-CoV-2.BioTech (Basel). 2023 Jun 11;12(2):46. doi: 10.3390/biotech12020046. BioTech (Basel). 2023. PMID: 37366794 Free PMC article.
-
Targeting aldose reductase using natural African compounds as promising agents for managing diabetic complications.Front Bioinform. 2025 Feb 6;5:1499255. doi: 10.3389/fbinf.2025.1499255. eCollection 2025. Front Bioinform. 2025. PMID: 39996053 Free PMC article.
-
Identification of Hypericin as a Candidate Repurposed Therapeutic Agent for COVID-19 and Its Potential Anti-SARS-CoV-2 Activity.Front Microbiol. 2022 Feb 10;13:828984. doi: 10.3389/fmicb.2022.828984. eCollection 2022. Front Microbiol. 2022. PMID: 35222340 Free PMC article.
References
-
- Al-Khodairy, F. M., Khan, M. K., Kunhi, M., Pulicat, M. S., Akhtar, S., & Arif, J. M. (2013). In Silico prediction of mechanism of Erysolin-induced apoptosis in human breast cancer cell lines. American Journal of Bioinformatics Research, 3, 62–71.
-
- Beck, B. R., Shin, B., Choi, Y., Park, S., & Kang, K. (2020). Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Computational and Structural Biotechnology Journal, 18, 784–790. 10.1016/j.csbj.2020.03.025 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous