

Inhibition of IRF5 hyperactivation protects from lupus onset and severity
- PMID: 32897883
- PMCID: PMC7685739
- DOI: 10.1172/JCI120288
Inhibition of IRF5 hyperactivation protects from lupus onset and severity
Abstract
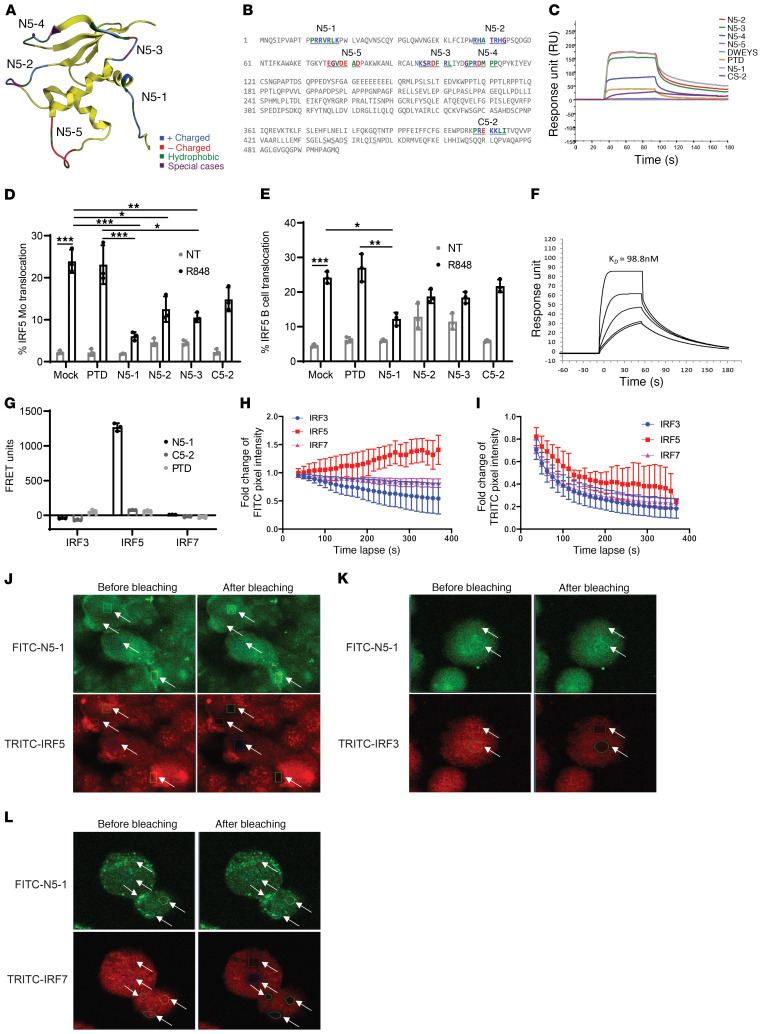
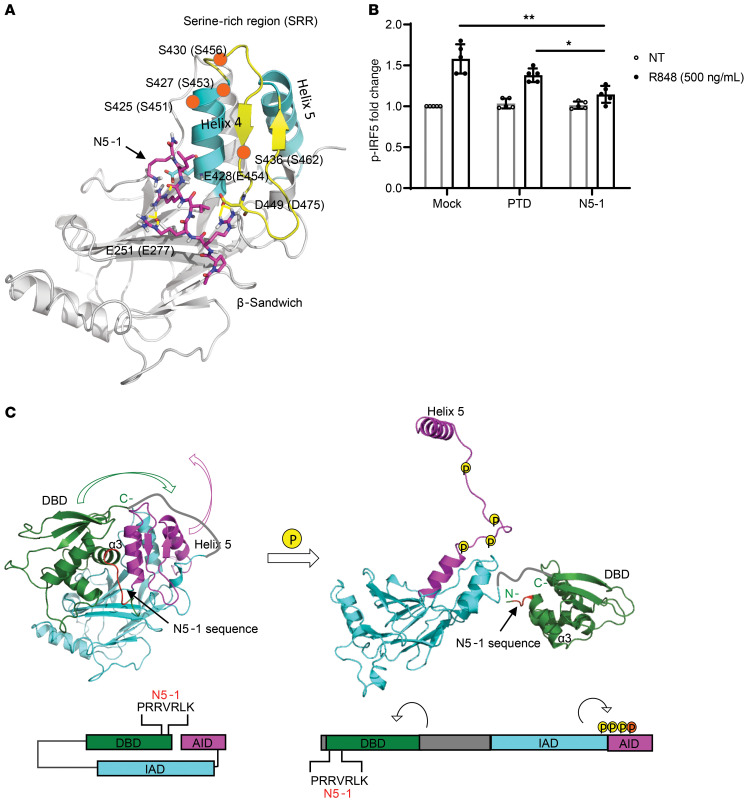
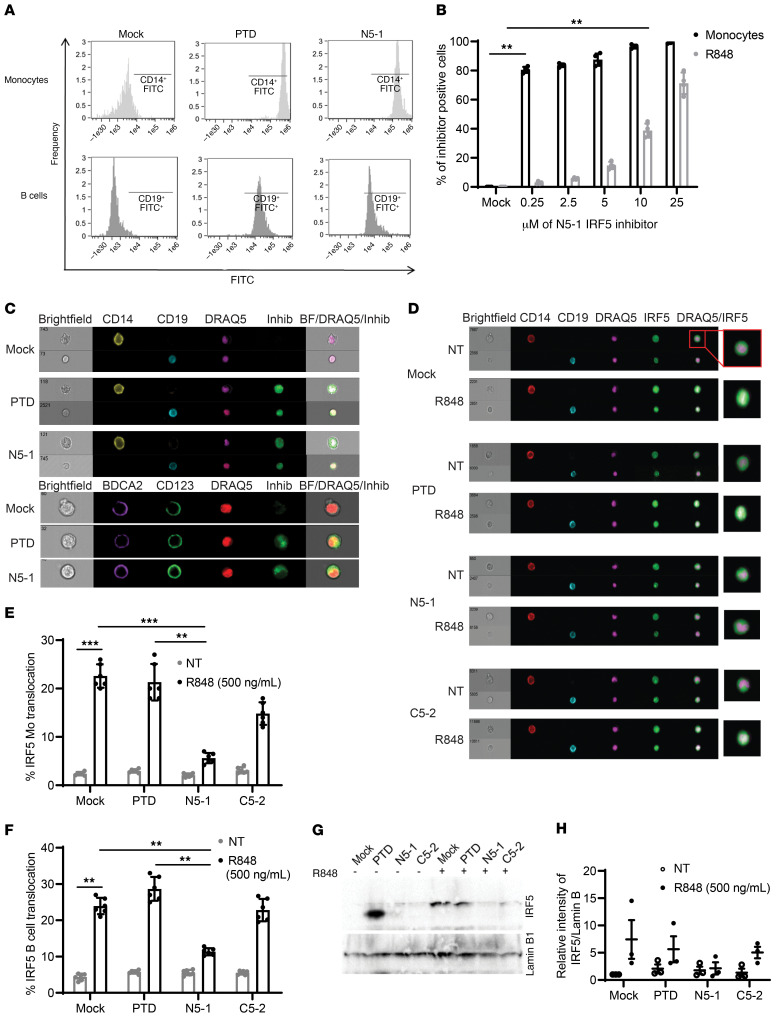
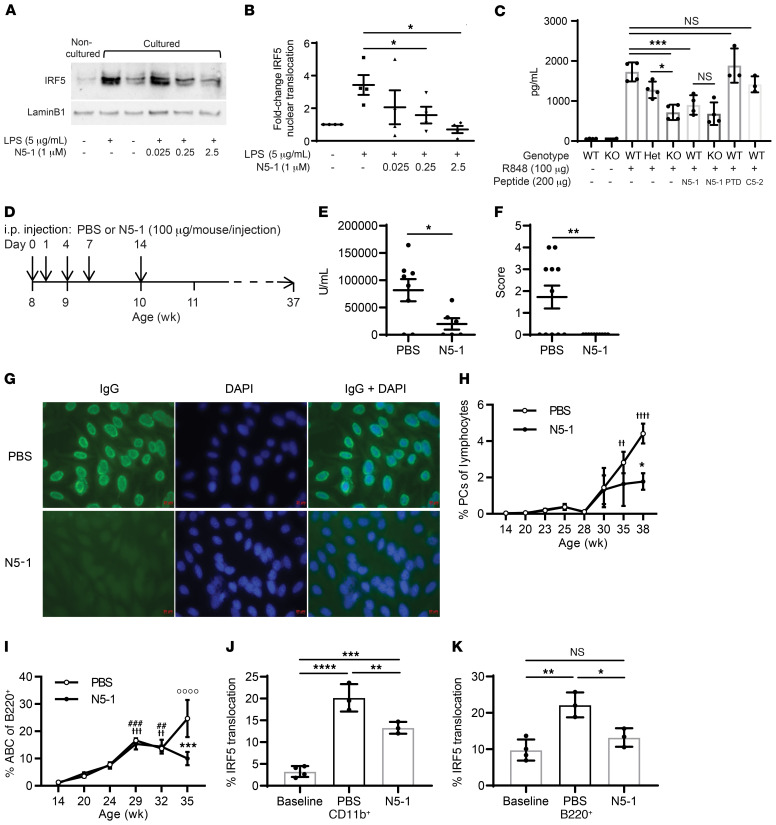
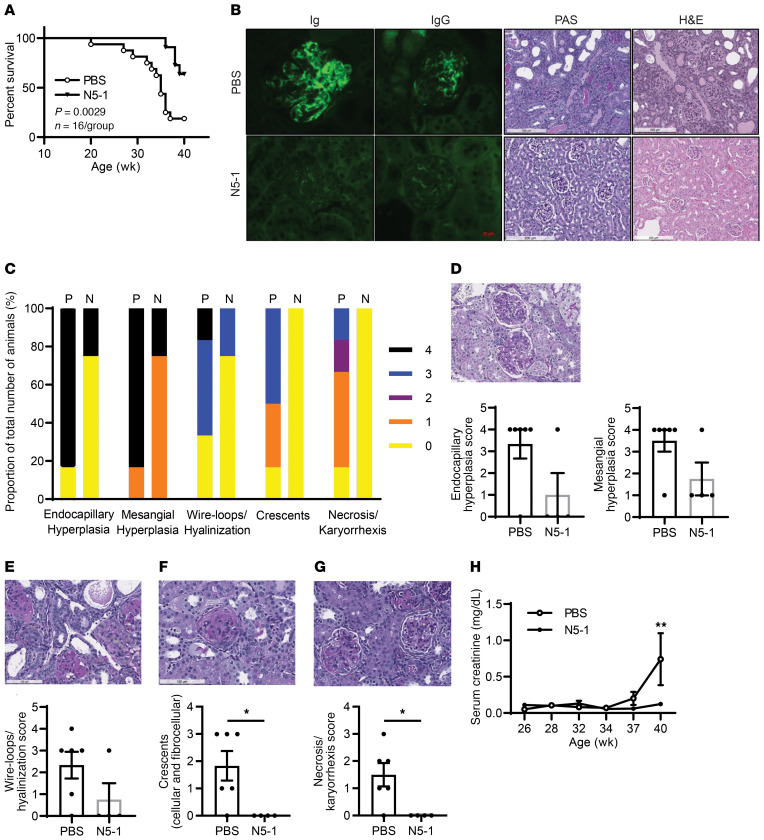
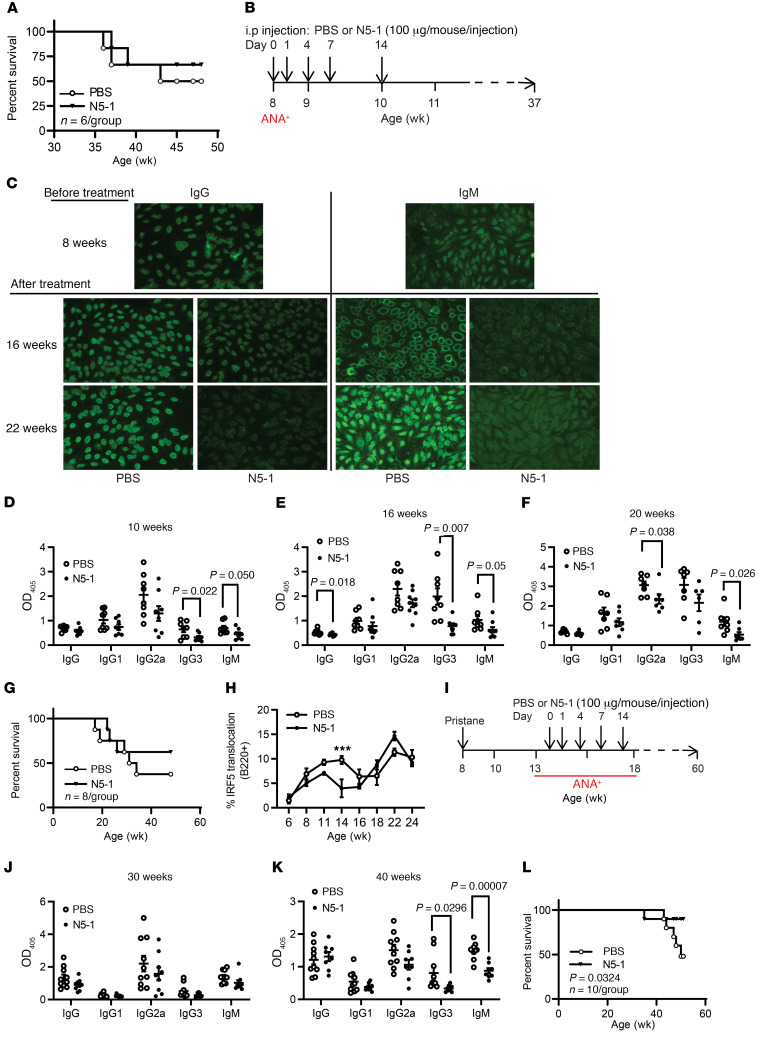
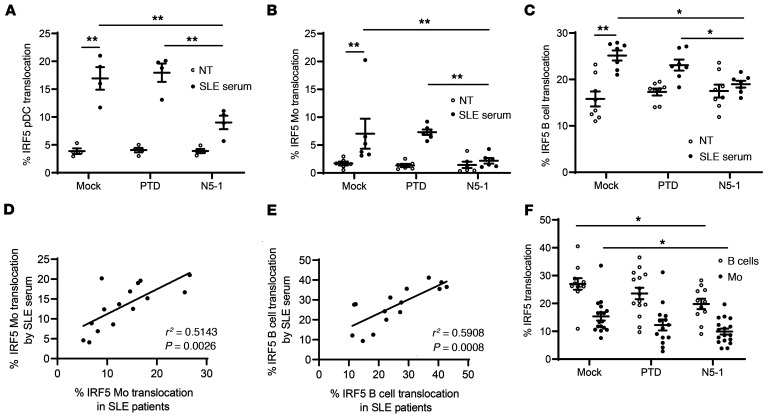
The transcription factor IFN regulatory factor 5 (IRF5) is a central mediator of innate and adaptive immunity. Genetic variations within IRF5 are associated with a risk of systemic lupus erythematosus (SLE), and mice lacking Irf5 are protected from lupus onset and severity, but how IRF5 functions in the context of SLE disease progression remains unclear. Using the NZB/W F1 model of murine lupus, we show that murine IRF5 becomes hyperactivated before clinical onset. In patients with SLE, IRF5 hyperactivation correlated with dsDNA titers. To test whether IRF5 hyperactivation is a targetable function, we developed inhibitors that are cell permeable, nontoxic, and selectively bind to the inactive IRF5 monomer. Preclinical treatment of NZB/W F1 mice with an inhibitor attenuated lupus pathology by reducing serum antinuclear autoantibodies, dsDNA titers, and the number of circulating plasma cells, which alleviated kidney pathology and improved survival. Clinical treatment of MRL/lpr and pristane-induced lupus mice with an inhibitor led to significant reductions in dsDNA levels and improved survival. In ex vivo human studies, the inhibitor blocked SLE serum-induced IRF5 activation and reversed basal IRF5 hyperactivation in SLE immune cells. We believe this study provides the first in vivo clinical support for treating patients with SLE with an IRF5 inhibitor.
Keywords: Autoimmune diseases; Autoimmunity; Immunology.
Conflict of interest statement
Figures

Comment in
-
IRF5 inhibitor shows promise in mouse models of SLE.Nat Rev Rheumatol. 2020 Dec;16(12):667. doi: 10.1038/s41584-020-00525-7. Nat Rev Rheumatol. 2020. PMID: 33024294 No abstract available.
References
-
- Ban T, Sato GR, Tamura T. Regulation and role of the transcription factor IRF5 in innate immune responses and systemic lupus erythematosus. Int Immunol. 2018;30(11):529–536. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
