

Targeting the Initiator Protease of the Classical Pathway of Complement Using Fragment-Based Drug Discovery
- PMID: 32899120
- PMCID: PMC7504721
- DOI: 10.3390/molecules25174016
Targeting the Initiator Protease of the Classical Pathway of Complement Using Fragment-Based Drug Discovery
Abstract
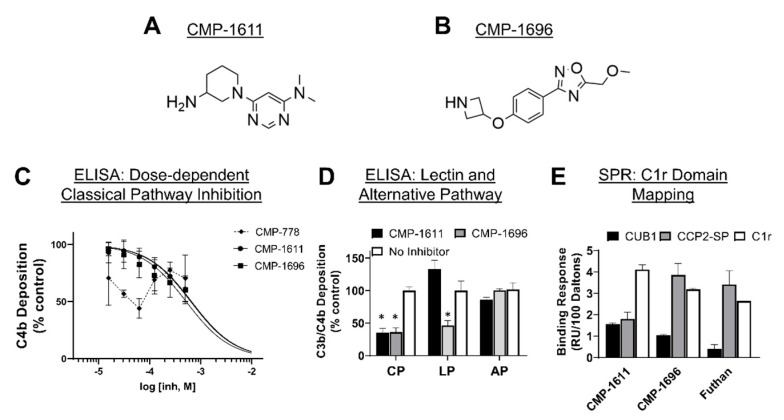
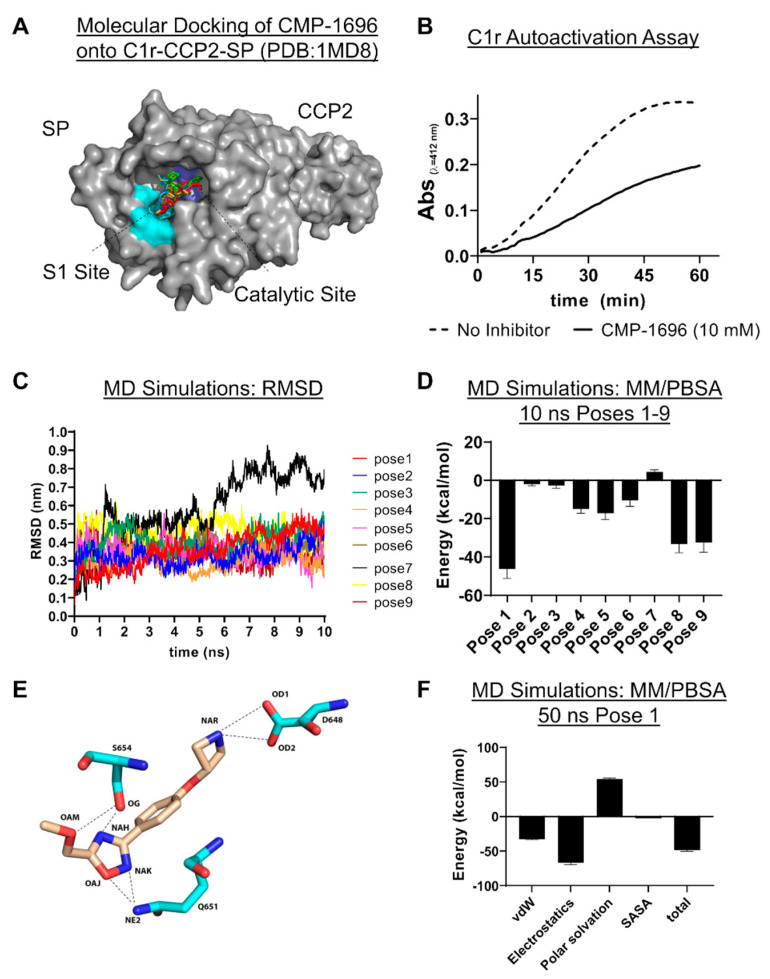
The initiating protease of the complement classical pathway, C1r, represents an upstream and pathway-specific intervention point for complement-related autoimmune and inflammatory diseases. Yet, C1r-targeted therapeutic development is currently underrepresented relative to other complement targets. In this study, we developed a fragment-based drug discovery approach using surface plasmon resonance (SPR) and molecular modeling to identify and characterize novel C1r-binding small-molecule fragments. SPR was used to screen a 2000-compound fragment library for binding to human C1r. This led to the identification of 24 compounds that bound C1r with equilibrium dissociation constants ranging between 160-1700 µM. Two fragments, termed CMP-1611 and CMP-1696, directly inhibited classical pathway-specific complement activation in a dose-dependent manner. CMP-1611 was selective for classical pathway inhibition, while CMP-1696 also blocked the lectin pathway but not the alternative pathway. Direct binding experiments mapped the CMP-1696 binding site to the serine protease domain of C1r and molecular docking and molecular dynamics studies, combined with C1r autoactivation assays, suggest that CMP-1696 binds within the C1r active site. The group of structurally distinct fragments identified here, along with the structure-activity relationship profiling of two lead fragments, form the basis for future development of novel high-affinity C1r-binding, classical pathway-specific, small-molecule complement inhibitors.
Keywords: complement inhibitors; fragment-based drug discovery; surface plasmon resonance.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
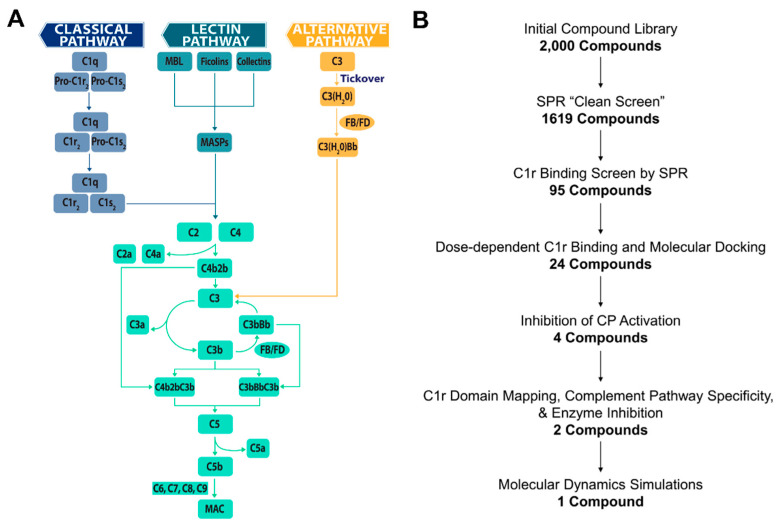
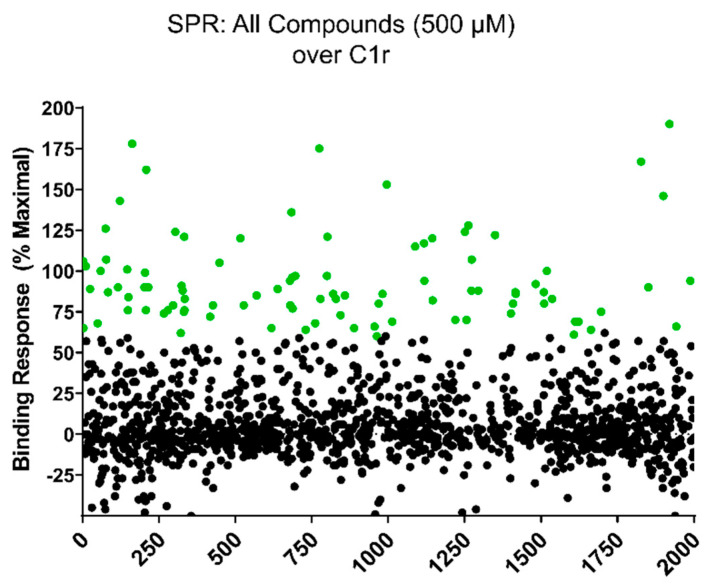
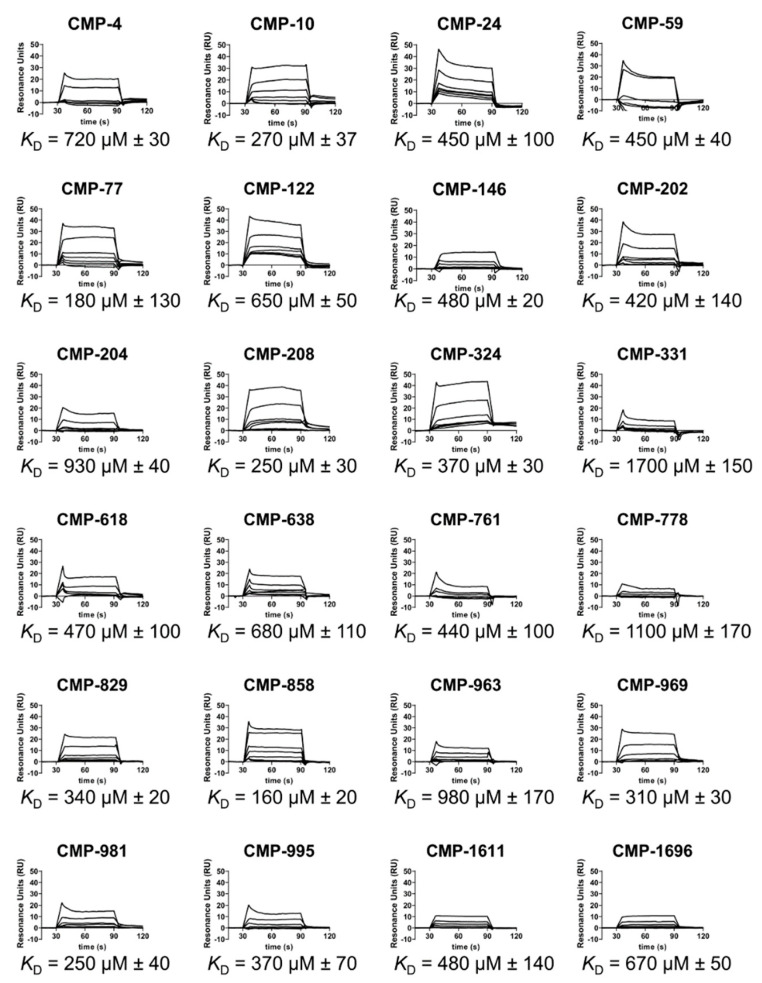
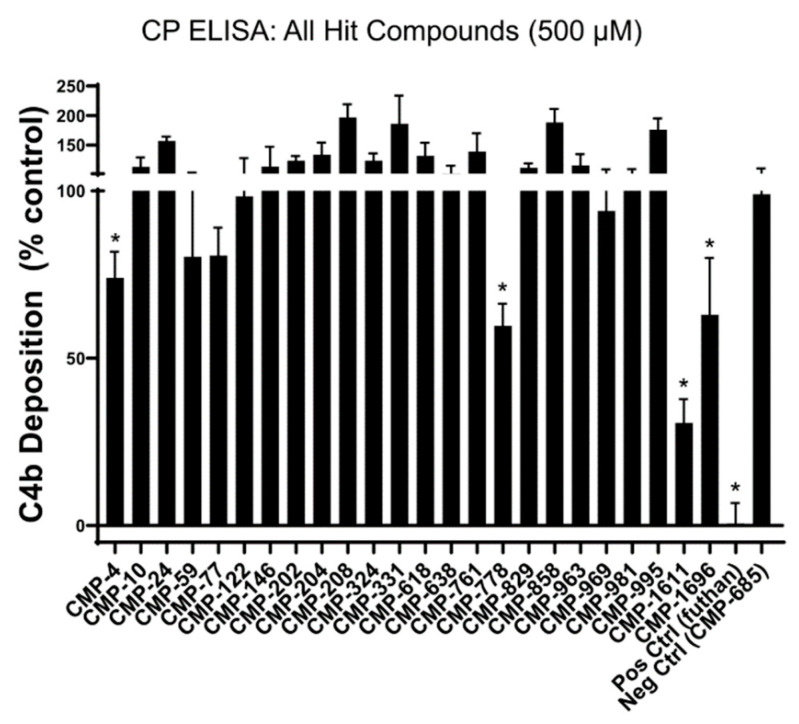
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
