

Targeting Alternative Splicing as a Potential Therapy for Episodic Ataxia Type 2
- PMID: 32899500
- PMCID: PMC7555146
- DOI: 10.3390/biomedicines8090332
Targeting Alternative Splicing as a Potential Therapy for Episodic Ataxia Type 2
Abstract
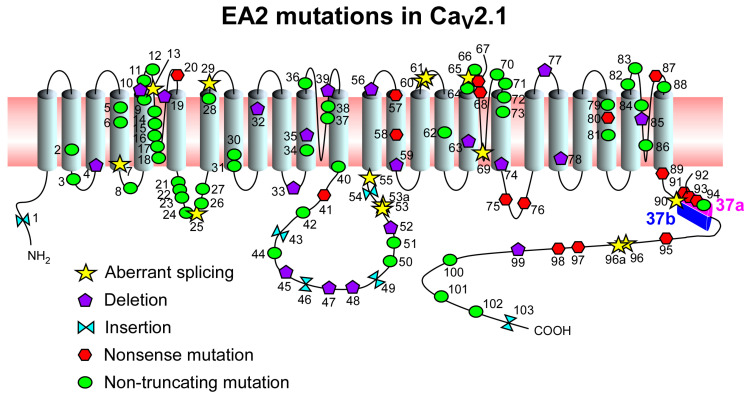
Episodic ataxia type 2 (EA2) is an autosomal dominant neurological disorder characterized by paroxysmal attacks of ataxia, vertigo, and nausea that usually last hours to days. It is caused by loss-of-function mutations in CACNA1A, the gene encoding the pore-forming α1 subunit of P/Q-type voltage-gated Ca2+ channels. Although pharmacological treatments, such as acetazolamide and 4-aminopyridine, exist for EA2, they do not reduce or control the symptoms in all patients. CACNA1A is heavily spliced and some of the identified EA2 mutations are predicted to disrupt selective isoforms of this gene. Modulating splicing of CACNA1A may therefore represent a promising new strategy to develop improved EA2 therapies. Because RNA splicing is dysregulated in many other genetic diseases, several tools, such as antisense oligonucleotides, trans-splicing, and CRISPR-based strategies, have been developed for medical purposes. Here, we review splicing-based strategies used for genetic disorders, including those for Duchenne muscular dystrophy, spinal muscular dystrophy, and frontotemporal dementia with Parkinsonism linked to chromosome 17, and discuss their potential applicability to EA2.
Keywords: CRISPR/Cas9; P/Q-type Ca2+ channels; SMaRT; alternative splicing; antisense oligonucleotides; episodic ataxia type 2.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
Similar articles
-
Functional implications of a novel EA2 mutation in the P/Q-type calcium channel.Ann Neurol. 2004 Aug;56(2):213-20. doi: 10.1002/ana.20169. Ann Neurol. 2004. PMID: 15293273
-
Epilepsy and episodic ataxia type 2: family study and review of the literature.J Neurol. 2021 Nov;268(11):4296-4302. doi: 10.1007/s00415-021-10555-0. Epub 2021 May 13. J Neurol. 2021. PMID: 33983550 Review.
-
Episodic Ataxia Type 2 – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY.2003 Feb 24 [updated 2015 Oct 15]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2003 Feb 24 [updated 2015 Oct 15]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301674 Free Books & Documents. Review.
-
Case Report: A Novel CACNA1A Mutation Caused Flunarizine-Responsive Type 2 Episodic Ataxia and Hemiplegic Migraine With Abnormal MRI of Cerebral White Matter.Front Neurol. 2022 May 23;13:899813. doi: 10.3389/fneur.2022.899813. eCollection 2022. Front Neurol. 2022. PMID: 35677330 Free PMC article.
-
Ubiquitin Ligase RNF138 Promotes Episodic Ataxia Type 2-Associated Aberrant Degradation of Human Cav2.1 (P/Q-Type) Calcium Channels.J Neurosci. 2017 Mar 1;37(9):2485-2503. doi: 10.1523/JNEUROSCI.3070-16.2017. Epub 2017 Feb 6. J Neurosci. 2017. PMID: 28167673 Free PMC article.
Cited by
-
The tetraspanin TSPAN5 regulates AMPAR exocytosis by interacting with the AP4 complex.Elife. 2023 Feb 16;12:e76425. doi: 10.7554/eLife.76425. Elife. 2023. PMID: 36795458 Free PMC article.
-
From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing.Front Neurol. 2021 Mar 2;12:639994. doi: 10.3389/fneur.2021.639994. eCollection 2021. Front Neurol. 2021. PMID: 33737904 Free PMC article. Review.
-
Genetic disorders of neurotransmitter release machinery.Front Synaptic Neurosci. 2023 Mar 31;15:1148957. doi: 10.3389/fnsyn.2023.1148957. eCollection 2023. Front Synaptic Neurosci. 2023. PMID: 37066095 Free PMC article. Review.
-
Long read sequencing reveals novel isoforms and insights into splicing regulation during cell state changes.BMC Genomics. 2022 Jan 10;23(1):42. doi: 10.1186/s12864-021-08261-2. BMC Genomics. 2022. PMID: 35012468 Free PMC article.
-
CACNA1A Mutations Causing Early Onset Ataxia: Profiling Clinical, Dysmorphic and Structural-Functional Findings.Int J Mol Sci. 2021 May 13;22(10):5180. doi: 10.3390/ijms22105180. Int J Mol Sci. 2021. PMID: 34068417 Free PMC article.
References
-
- Mantuano E., Romano S., Veneziano L., Gellera C., Castellotti B., Caimi S., Testa D., Estienne M., Zorzi G., Bugiani M., et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J. Neurol. Sci. 2010;291:30–36. doi: 10.1016/j.jns.2010.01.010. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
