

Hidden dynamic signatures drive substrate selectivity in the disordered phosphoproteome
- PMID: 32900925
- PMCID: PMC7519349
- DOI: 10.1073/pnas.1921473117
Hidden dynamic signatures drive substrate selectivity in the disordered phosphoproteome
Abstract
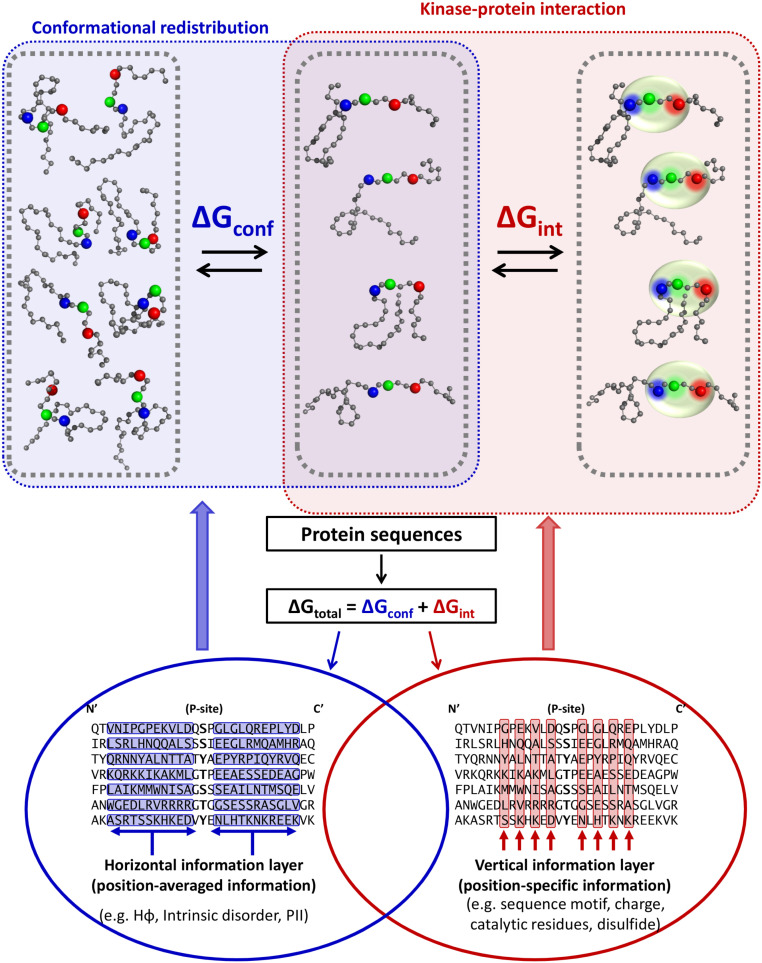
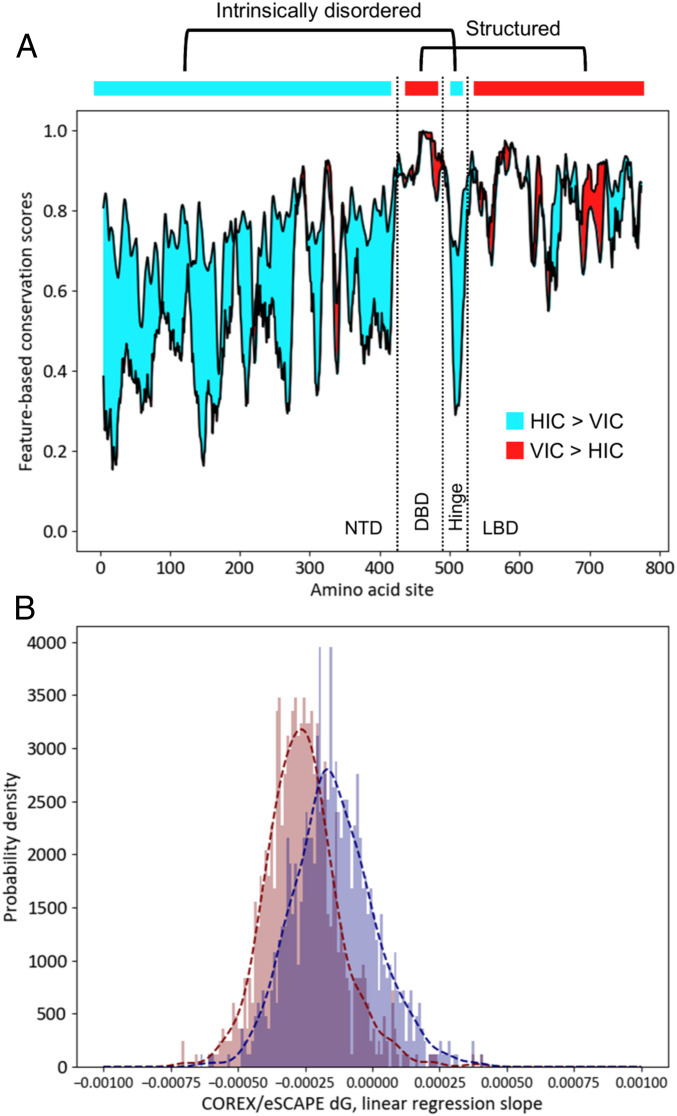
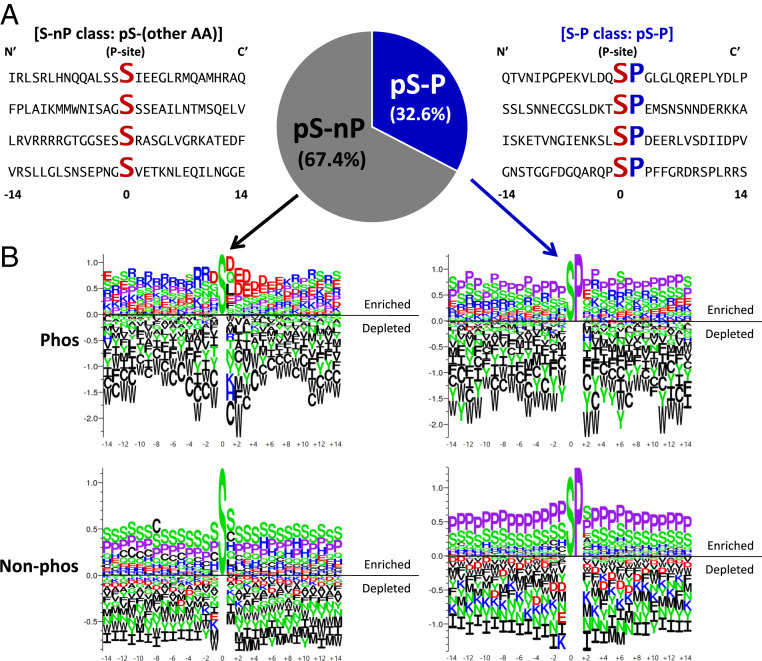
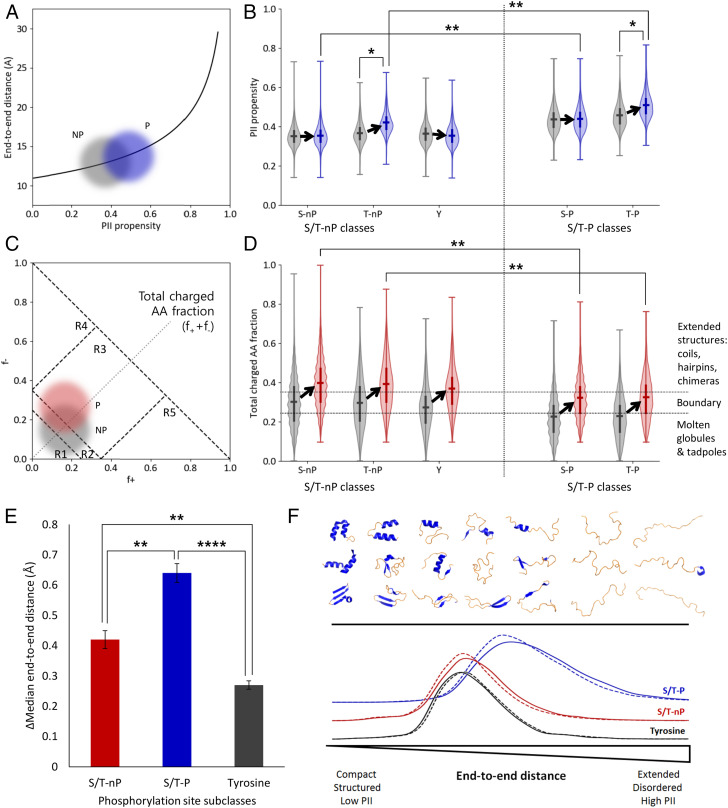
Phosphorylation sites are hyperabundant in the eukaryotic disordered proteome, suggesting that conformational fluctuations play a major role in determining to what extent a kinase interacts with a particular substrate. In biophysical terms, substrate selectivity may be determined not just by the structural-chemical complementarity between the kinase and its protein substrates but also by the free energy difference between the conformational ensembles that are, or are not, recognized by the kinase. To test this hypothesis, we developed a statistical-thermodynamics-based informatics framework, which allows us to probe for the contribution of equilibrium fluctuations to phosphorylation, as evaluated by the ability to predict Ser/Thr/Tyr phosphorylation sites in the disordered proteome. Essential to this framework is a decomposition of substrate sequence information into two types: vertical information encoding conserved kinase specificity motifs and horizontal information encoding substrate conformational equilibrium that is embedded, but often not apparent, within position-specific conservation patterns. We find not only that conformational fluctuations play a major role but also that they are the dominant contribution to substrate selectivity. In fact, the main substrate classifier distinguishing selectivity is the magnitude of change in local compaction of the disordered chain upon phosphorylation of these mostly singly phosphorylated sites. In addition to providing fundamental insights into the consequences of phosphorylation across the proteome, our approach provides a statistical-thermodynamic strategy for partitioning any sequence-based search into contributions from structural-chemical complementarity and those from changes in conformational equilibrium.
Keywords: cellular signaling; conformational equilibrium; intrinsic disorder; local unfolding; protein ensemble.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
Similar articles
-
Conformational ensembles of the human intrinsically disordered proteome.Nature. 2024 Feb;626(8000):897-904. doi: 10.1038/s41586-023-07004-5. Epub 2024 Jan 31. Nature. 2024. PMID: 38297118
-
The entropic force generated by intrinsically disordered segments tunes protein function.Nature. 2018 Nov;563(7732):584-588. doi: 10.1038/s41586-018-0699-5. Epub 2018 Nov 12. Nature. 2018. PMID: 30420606 Free PMC article.
-
Large-scale identification of phosphorylation sites for profiling protein kinase selectivity.J Proteome Res. 2014 Jul 3;13(7):3410-9. doi: 10.1021/pr500319y. Epub 2014 Jun 4. J Proteome Res. 2014. PMID: 24869485
-
Ligand interactions and the protein order-disorder energetic continuum.Semin Cell Dev Biol. 2020 Mar;99:78-85. doi: 10.1016/j.semcdb.2018.05.007. Epub 2018 Jun 5. Semin Cell Dev Biol. 2020. PMID: 29753880 Review.
-
Highly Disordered Proteins in Prostate Cancer.Curr Protein Pept Sci. 2017;18(5):453-481. doi: 10.2174/1389203717666161028145848. Curr Protein Pept Sci. 2017. PMID: 27804860 Review.
Cited by
-
The immune response to RNA suppresses nucleic acid synthesis by limiting ribose 5-phosphate.EMBO J. 2024 Jul;43(13):2636-2660. doi: 10.1038/s44318-024-00100-w. Epub 2024 May 22. EMBO J. 2024. PMID: 38778156 Free PMC article.
-
Conserved allosteric ensembles in disordered proteins using TROSY/anti-TROSY R2-filtered spectroscopy.Biophys J. 2021 Jun 15;120(12):2498-2510. doi: 10.1016/j.bpj.2021.04.017. Epub 2021 Apr 24. Biophys J. 2021. PMID: 33901472 Free PMC article.
-
Mechanotransduction as a major driver of cell behaviour: mechanisms, and relevance to cell organization and future research.Open Biol. 2021 Nov;11(11):210256. doi: 10.1098/rsob.210256. Epub 2021 Nov 10. Open Biol. 2021. PMID: 34753321 Free PMC article. Review.
-
Evolutionary crossroads of cell signaling: PP1 and PP2A substrate sites in intrinsically disordered regions.Biochem Soc Trans. 2021 Jun 30;49(3):1065-1074. doi: 10.1042/BST20200175. Biochem Soc Trans. 2021. PMID: 34100859 Free PMC article. Review.
-
Molecular dynamics simulations of intrinsically disordered protein regions enable biophysical interpretation of variant effect predictors.bioRxiv [Preprint]. 2025 May 12:2025.05.07.652723. doi: 10.1101/2025.05.07.652723. bioRxiv. 2025. PMID: 40462930 Free PMC article. Preprint.
References
-
- Collins M. O., Yu L., Choudhary J. S., Analysis of protein phosphorylation on a proteome-scale. Proteomics 7, 2751–2768 (2007). - PubMed
-
- Deribe Y. L., Pawson T., Dikic I., Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672 (2010). - PubMed
-
- Humphrey S. J., James D. E., Mann M., Protein phosphorylation: A major switch mechanism for metabolic regulation. Trends Endocrinol. Metab. 26, 676–687 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
