

Large scale in silico characterization of repeat expansion variation in human genomes
- PMID: 32901039
- PMCID: PMC7479135
- DOI: 10.1038/s41597-020-00633-9
Large scale in silico characterization of repeat expansion variation in human genomes
Abstract
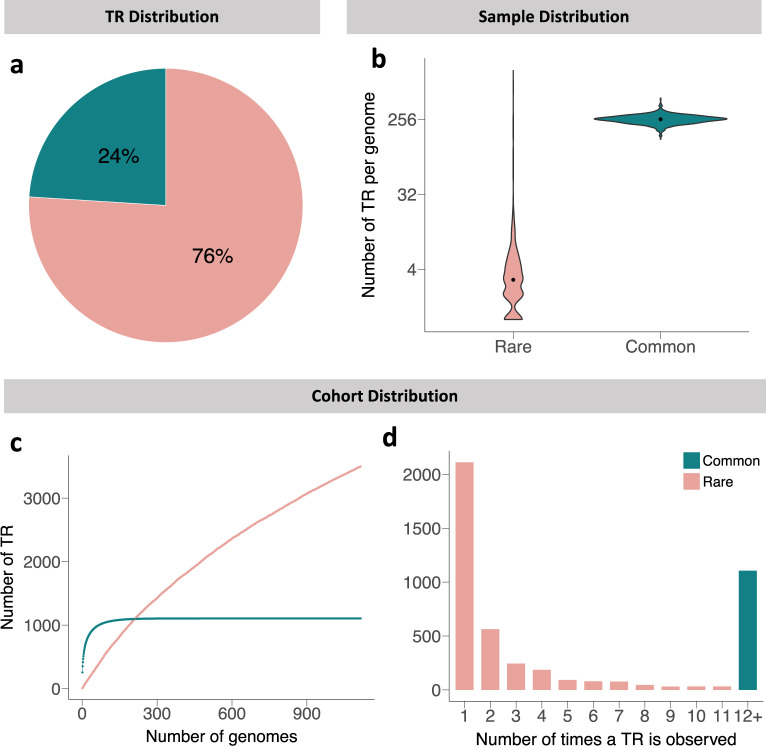
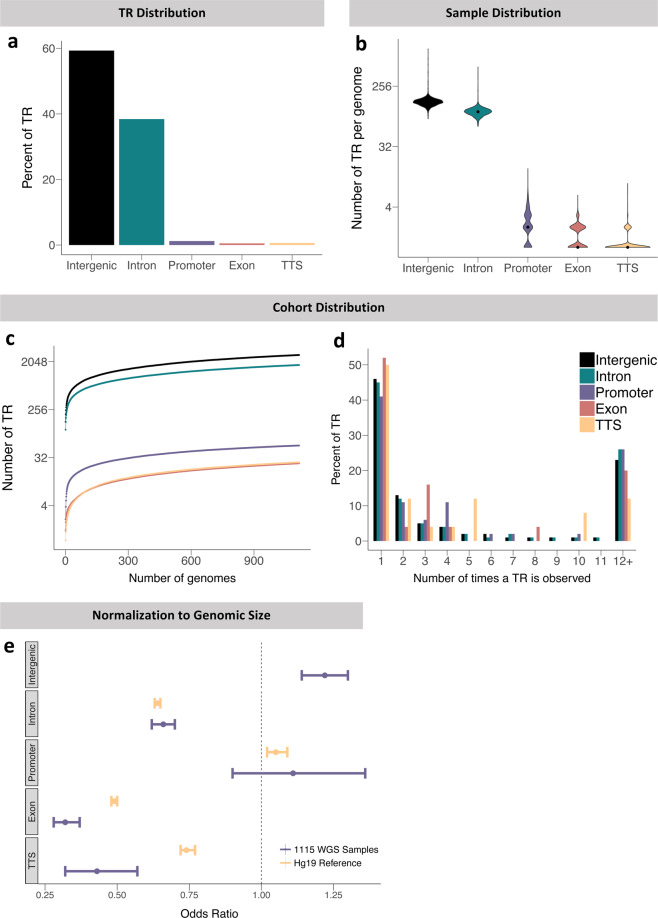
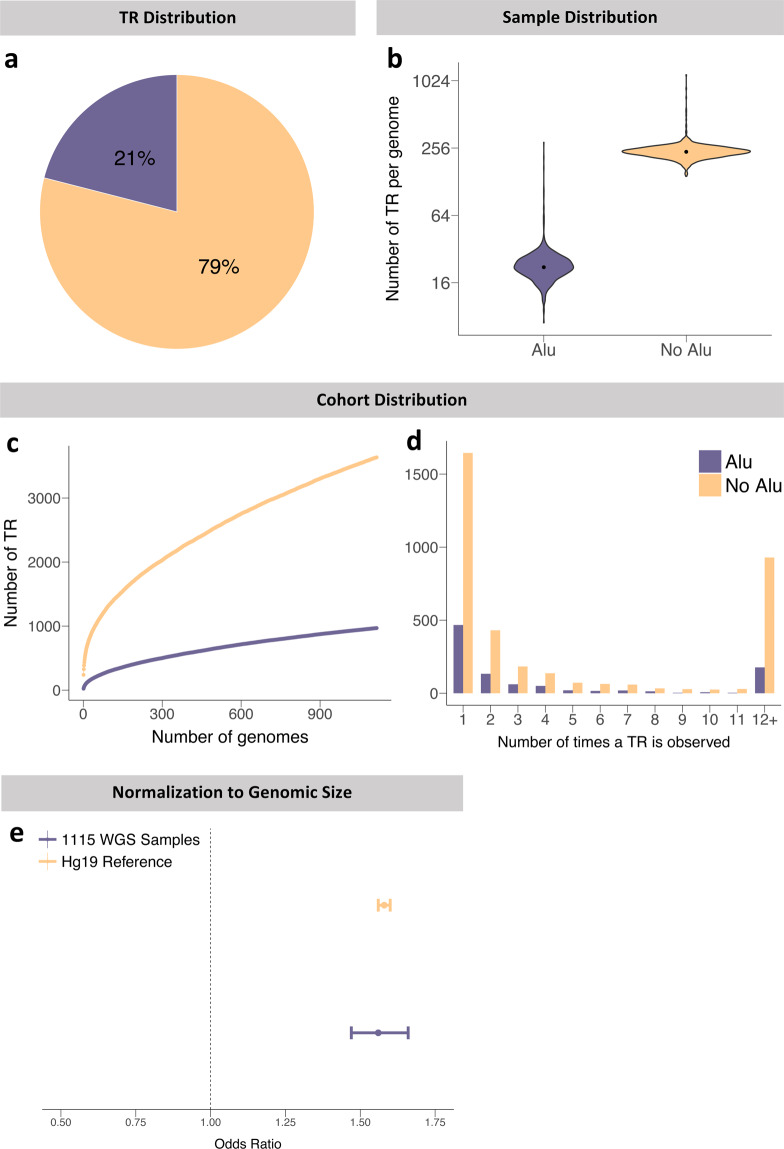
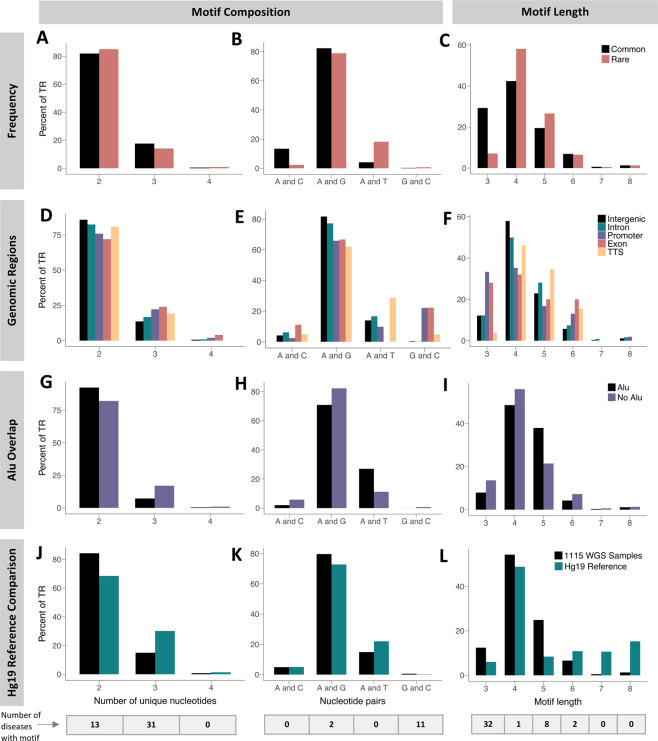
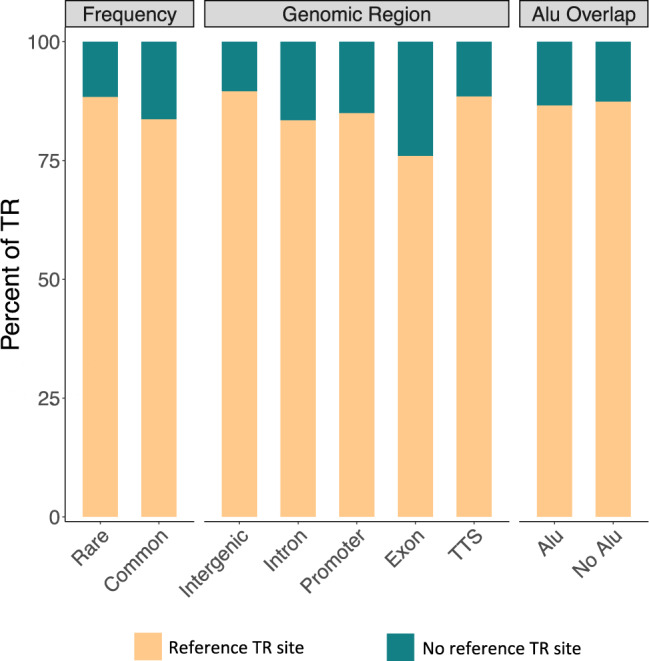
Significant progress has been made in elucidating single nucleotide polymorphism diversity in the human population. However, the majority of the variation space in the genome is structural and remains partially elusive. One form of structural variation is tandem repeats (TRs). Expansion of TRs are responsible for over 40 diseases, but we hypothesize these represent only a fraction of the pathogenic repeat expansions that exist. Here we characterize long or expanded TR variation in 1,115 human genomes as well as a replication cohort of 2,504 genomes, identified using ExpansionHunter Denovo. We found that individual genomes typically harbor several rare, large TRs, generally in non-coding regions of the genome. We noticed that these large TRs are enriched in their proximity to Alu elements. The vast majority of these large TRs seem to be expansions of smaller TRs that are already present in the reference genome. We are providing this TR profile as a resource for comparison to undiagnosed rare disease genomes in order to detect novel disease-causing repeat expansions.
Conflict of interest statement
E.D. and M.A.E. are employees of Illumina, Inc., a public company that develops and markets systems for genetic analysis.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
