

A classic variant of Fabry disease in a family with the M296I late-onset variant
- PMID: 32902816
- PMCID: PMC7829310
- DOI: 10.1007/s13730-020-00527-0
A classic variant of Fabry disease in a family with the M296I late-onset variant
Abstract
Fabry disease is an X-linked recessive disease of glycosphingolipid metabolism caused by deficiency or reduced activity of α-galactosidase A. Fabry disease phenotypes are known to consist of a classic variant and a late-onset variant. In patients with Fabry disease, the phenotype is generally considered to be defined (at least partially) by the genotype. However, patients with the classic variant have been encountered in families with mutations that are expected to produce the late-onset variant. Here, we describe a 4-year-old boy with a classic variant of Fabry disease in a family with the M296I late-onset variant. The patient's grandfather, mother, and aunt experienced late-onset disease, characteristic of the M296I variant. Conversely, the patient experienced typical disease symptoms in childhood. He had symptoms of hypohidrosis and associated heat accumulation. He cried at night due to the occurrence of severe acroparaesthesia. This symptom became more pronounced in warmer climates. Although the patient's family had a late-onset variant mutation of Fabry disease, we determined that the patient's symptoms were similar to those of classic Fabry disease. Therefore, the patient began enzyme replacement therapy, which alleviated his symptoms. Notably, enzyme replacement therapy led to rapid improvement of the patient's subjective symptoms. Thus, we presumed that the patient's symptoms supported a diagnosis of classic Fabry disease. These findings suggest that childhood symptoms may occur in patients with Fabry disease, even in families with late-onset variant mutations. The genotype-phenotype correlation in Fabry disease remains controversial.
Keywords: Classic variant; Fabry disease; Genotype; Late-onset variant; M296I mutation; Phenotype.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
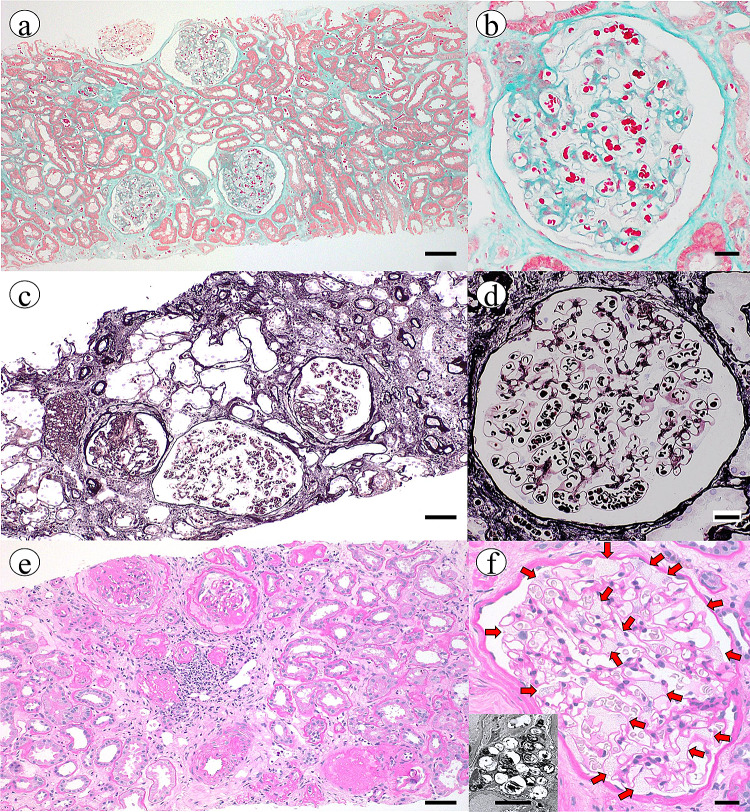
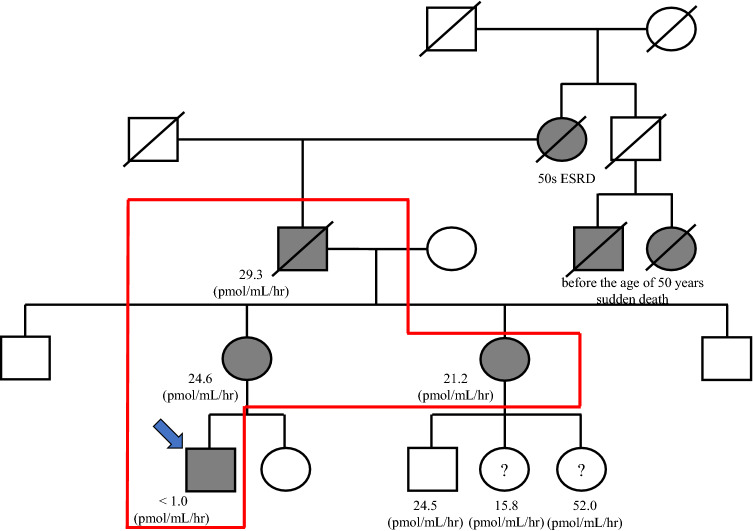
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
