

Ape Origins of Human Malaria
- PMID: 32905751
- PMCID: PMC7643433
- DOI: 10.1146/annurev-micro-020518-115628
Ape Origins of Human Malaria
Abstract
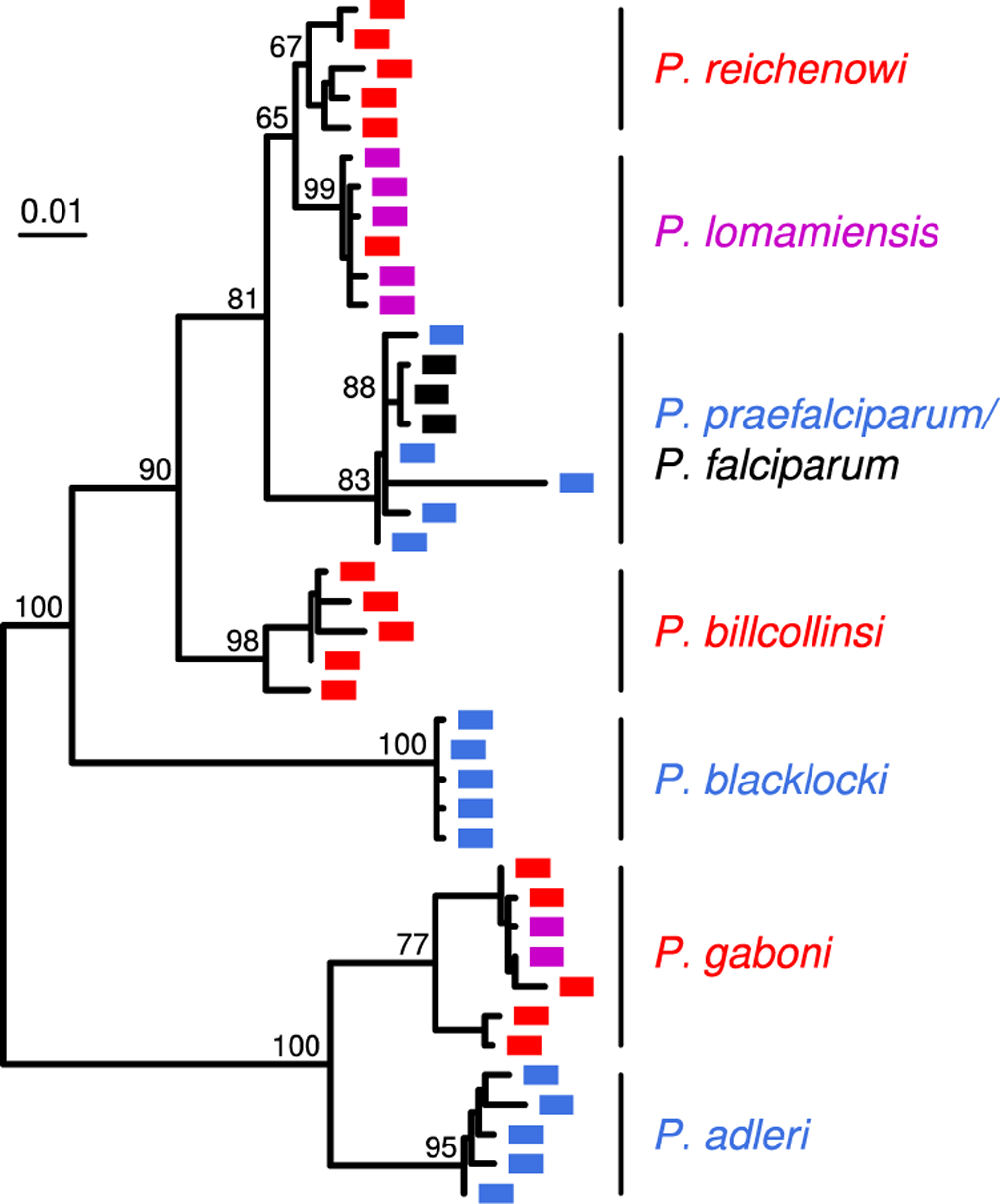
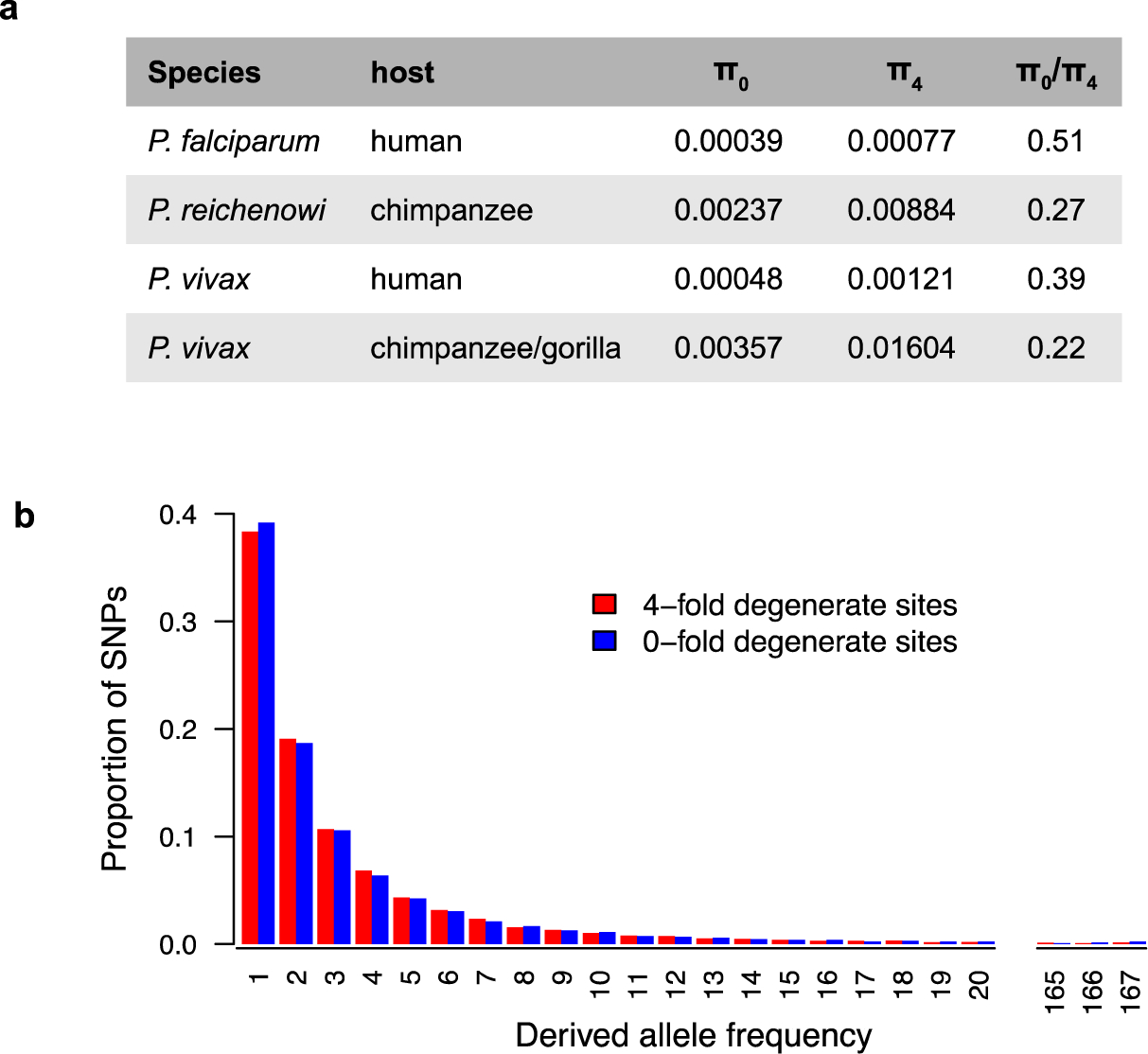
African apes harbor at least twelve Plasmodium species, some of which have been a source of human infection. It is now well established that Plasmodium falciparum emerged following the transmission of a gorilla parasite, perhaps within the last 10,000 years, while Plasmodium vivax emerged earlier from a parasite lineage that infected humans and apes in Africa before the Duffy-negative mutation eliminated the parasite from humans there. Compared to their ape relatives, both human parasites have greatly reduced genetic diversity and an excess of nonsynonymous mutations, consistent with severe genetic bottlenecks followed by rapid population expansion. A putative new Plasmodium species widespread in chimpanzees, gorillas, and bonobos places the origin of Plasmodium malariae in Africa. Here, we review what is known about the origins and evolutionary history of all human-infective Plasmodium species, the time and circumstances of their emergence, and the diversity, host specificity, and zoonotic potential of their ape counterparts.
Keywords: Plasmodium; chimpanzee; cross species transmission; evolution; gorilla; interspecies gene transfer; malaria.
Figures





References
-
- 2014. Severe malaria. Trop Med Int Health 19 Suppl:7–131. World Health Organ. - PubMed
-
- Malar. Genom. Epidemiol. Netw. 2016. Pf3k pilot data release 5. MalariaGEN https://www.malariagen.net/data/pf3k-5
-
- World Health Organ. 2019. World Malaria Report 2019, Geneva: World Health Organ.
-
- Adler S 1923. Malaria in chimpanzees in Sierra Leone. Annals of Tropical Medicine & Parasitology 17:13–8
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
