

"Repair Me if You Can": Membrane Damage, Response, and Control from the Viral Perspective
- PMID: 32906744
- PMCID: PMC7564661
- DOI: 10.3390/cells9092042
"Repair Me if You Can": Membrane Damage, Response, and Control from the Viral Perspective
Abstract
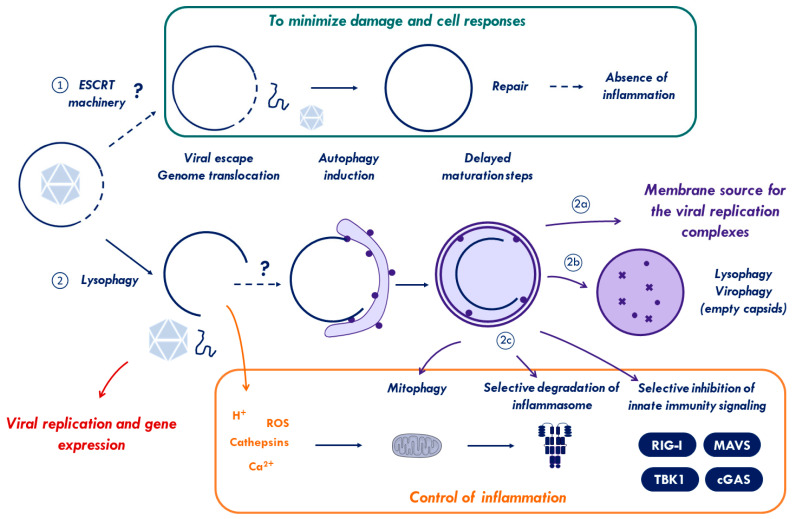
Cells are constantly challenged by pathogens (bacteria, virus, and fungi), and protein aggregates or chemicals, which can provoke membrane damage at the plasma membrane or within the endo-lysosomal compartments. Detection of endo-lysosomal rupture depends on a family of sugar-binding lectins, known as galectins, which sense the abnormal exposure of glycans to the cytoplasm upon membrane damage. Galectins in conjunction with other factors orchestrate specific membrane damage responses such as the recruitment of the endosomal sorting complex required for transport (ESCRT) machinery to either repair damaged membranes or the activation of autophagy to remove membrane remnants. If not controlled, membrane damage causes the release of harmful components including protons, reactive oxygen species, or cathepsins that will elicit inflammation. In this review, we provide an overview of current knowledge on membrane damage and cellular responses. In particular, we focus on the endo-lysosomal damage triggered by non-enveloped viruses (such as adenovirus) and discuss viral strategies to control the cellular membrane damage response. Finally, we debate the link between autophagy and inflammation in this context and discuss the possibility that virus induced autophagy upon entry limits inflammation.
Keywords: ESCRT machinery; adenovirus; antiviral autophagy; bacterial invasion; galectin; inflammation; interferon; lysophagy; membrane damage; virus entry.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
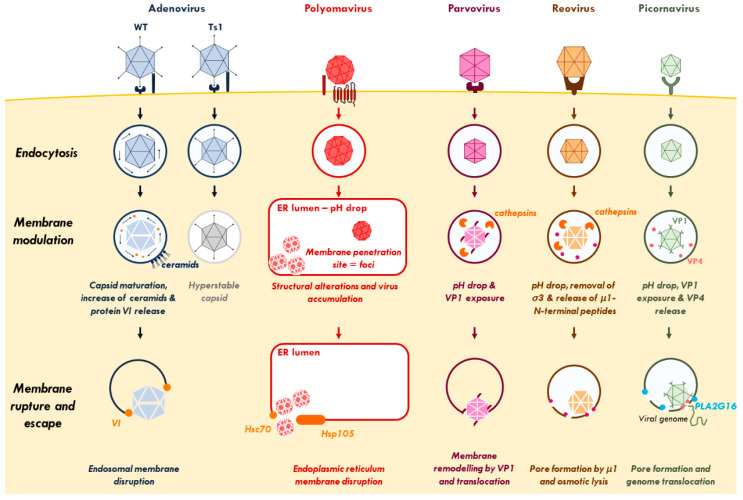
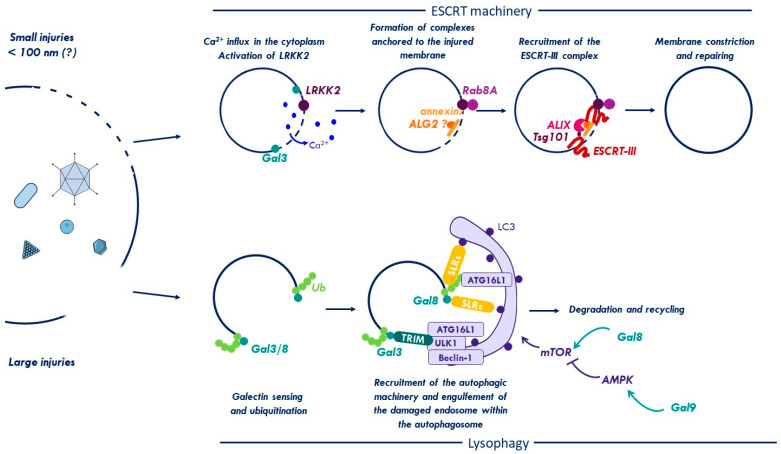
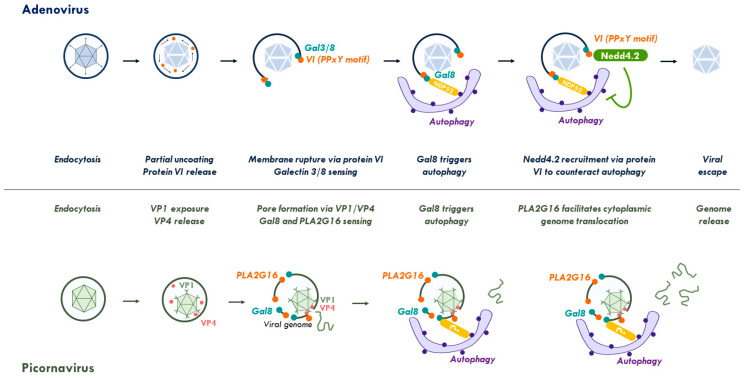
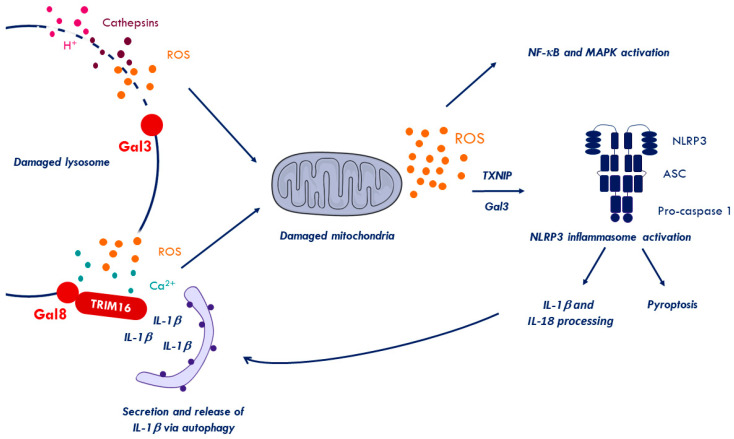
References
-
- Montespan C., Marvin S.A., Austin S., Burrage A.M., Roger B., Rayne F., Faure M., Campell E.M., Schneider C., Reimer R., et al. Multi-layered control of Galectin-8 mediated autophagy during adenovirus cell entry through a conserved PPxY motif in the viral capsid. PLoS Pathog. 2017;13 doi: 10.1371/journal.ppat.1006217. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
