

Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities
- PMID: 32906785
- PMCID: PMC7563346
- DOI: 10.3390/cells9092044
Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities
Abstract
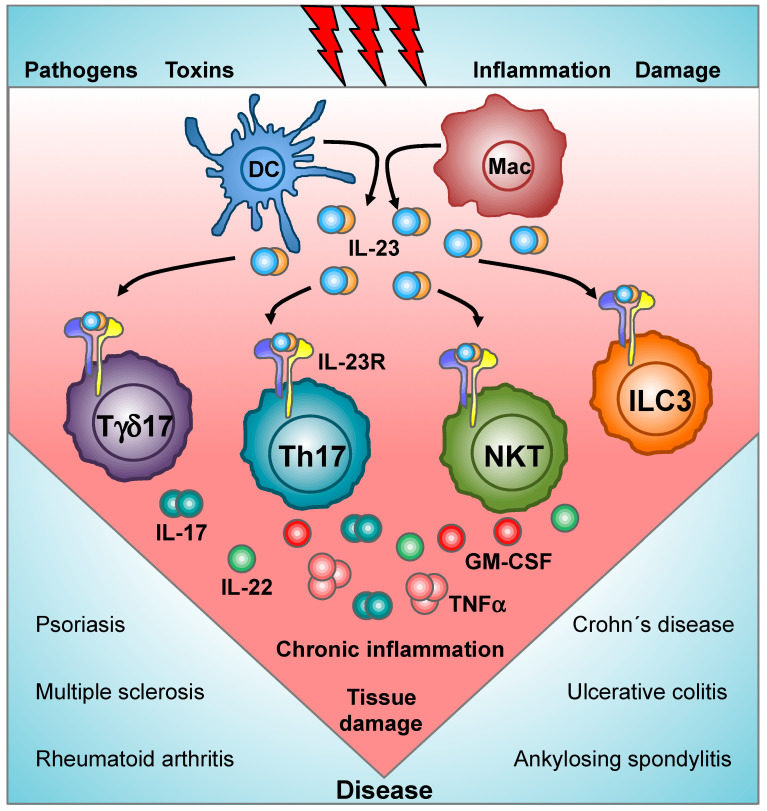
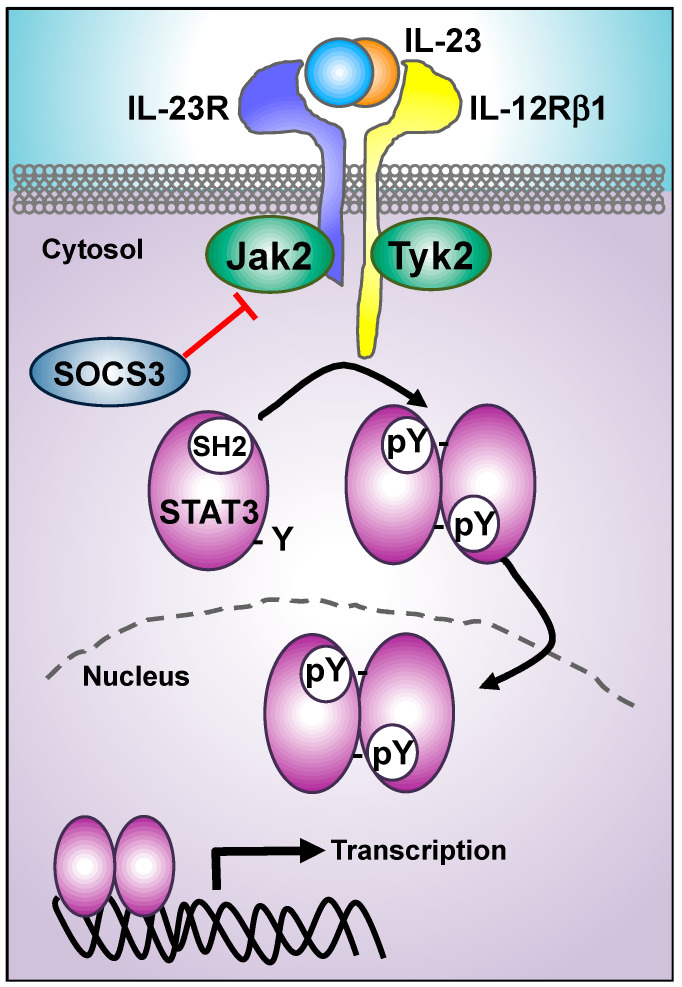
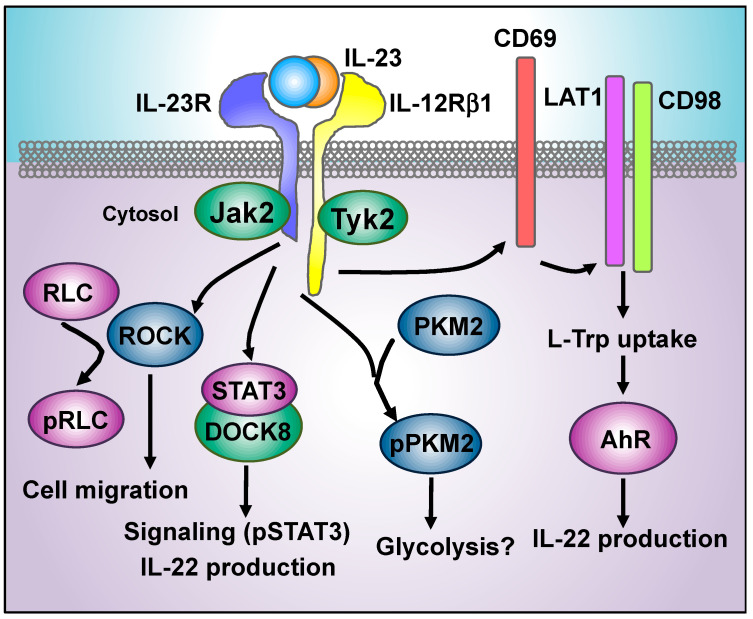
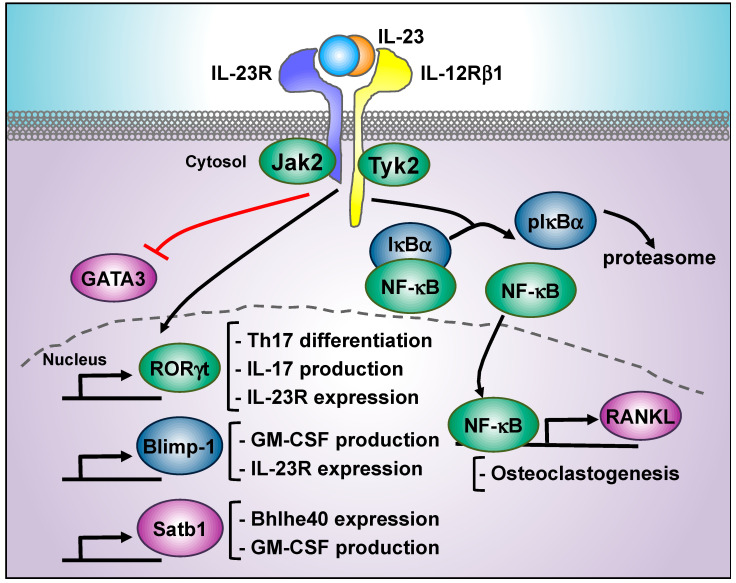
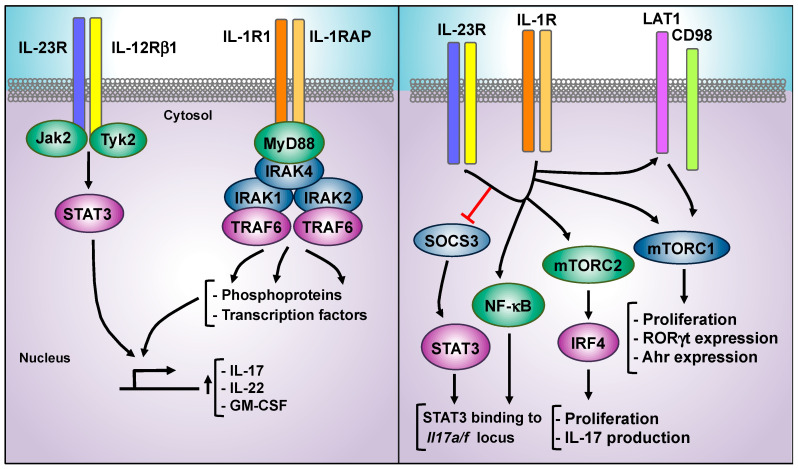
The interleukin 23 (IL-23) is a key pro-inflammatory cytokine in the development of chronic inflammatory diseases, such as psoriasis, inflammatory bowel diseases, multiple sclerosis, or rheumatoid arthritis. The pathological consequences of excessive IL-23 signaling have been linked to its ability to promote the production of inflammatory mediators, such as IL-17, IL-22, granulocyte-macrophage colony-stimulating (GM-CSF), or the tumor necrosis factor (TNFα) by target populations, mainly Th17 and IL-17-secreting TCRγδ cells (Tγδ17). Due to their pivotal role in inflammatory diseases, IL-23 and its downstream effector molecules have emerged as attractive therapeutic targets, leading to the development of neutralizing antibodies against IL-23 and IL-17 that have shown efficacy in different inflammatory diseases. Despite the success of monoclonal antibodies, there are patients that show no response or partial response to these treatments. Thus, effective therapies for inflammatory diseases may require the combination of multiple immune-modulatory drugs to prevent disease progression and to improve quality of life. Alternative strategies aimed at inhibiting intracellular signaling cascades using small molecule inhibitors or interfering peptides have not been fully exploited in the context of IL-23-mediated diseases. In this review, we discuss the current knowledge about proximal signaling events triggered by IL-23 upon binding to its membrane receptor to bring to the spotlight new opportunities for therapeutic intervention in IL-23-mediated pathologies.
Keywords: IL-23; autoimmunity; inflammatory disease; interleukin 17; interleukin 23; multiple sclerosis; psoriasis; signaling.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the writing of the manuscript, or in the decision to publish the results.
Figures
References
-
- Oppmann B., Lesley R., Blom B., Timans J.C., Xu Y., Hunte B., Vega F., Yu N., Wang J., Singh K., et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/S1074-7613(00)00070-4. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
