

Nucleotide Loading Modes of Human RNA Polymerase II as Deciphered by Molecular Simulations
- PMID: 32906795
- PMCID: PMC7565877
- DOI: 10.3390/biom10091289
Nucleotide Loading Modes of Human RNA Polymerase II as Deciphered by Molecular Simulations
Abstract
Mapping the route of nucleoside triphosphate (NTP) entry into the sequestered active site of RNA polymerase (RNAP) has major implications for elucidating the complete nucleotide addition cycle. Constituting a dichotomy that remains to be resolved, two alternatives, direct NTP delivery via the secondary channel (CH2) or selection to downstream sites in the main channel (CH1) prior to catalysis, have been proposed. In this study, accelerated molecular dynamics simulations of freely diffusing NTPs about RNAPII were applied to refine the CH2 model and uncover atomic details on the CH1 model that previously lacked a persuasive structural framework to illustrate its mechanism of action. Diffusion and binding of NTPs to downstream DNA, and the transfer of a preselected NTP to the active site, are simulated for the first time. All-atom simulations further support that CH1 loading is transcription factor IIF (TFIIF) dependent and impacts catalytic isomerization. Altogether, the alternative nucleotide loading systems may allow distinct transcriptional landscapes to be expressed.
Keywords: RNA polymerase; TFIIF; diffusion; downstream bubble; entry; loading; main channel; nucleoside triphosphate; secondary channel; tertiary channel.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


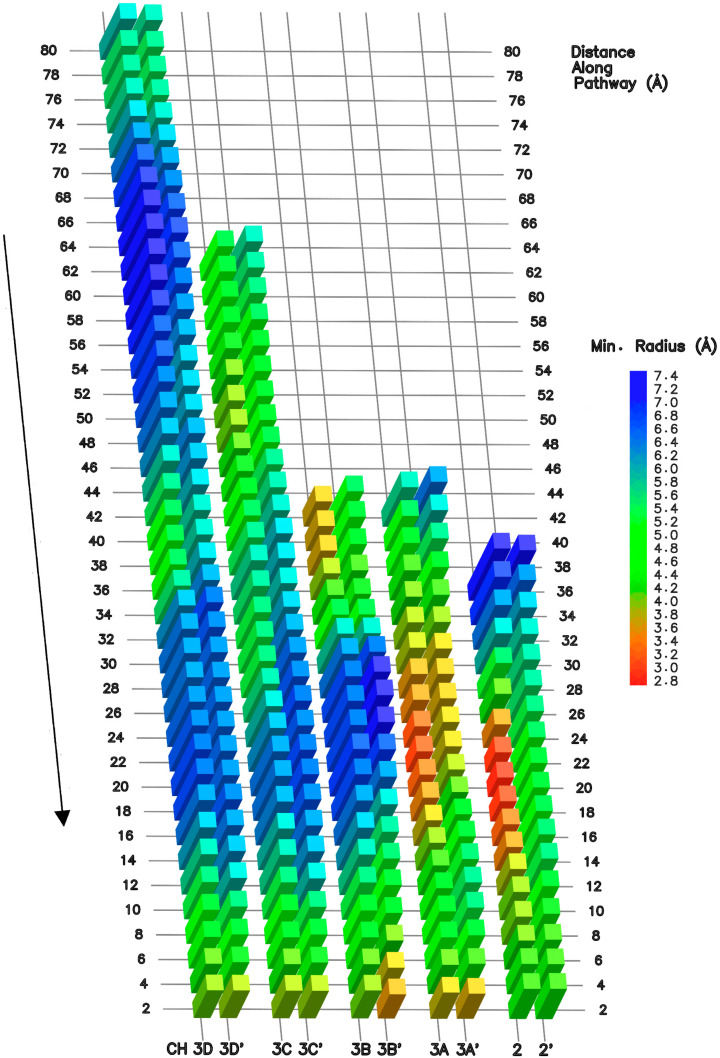
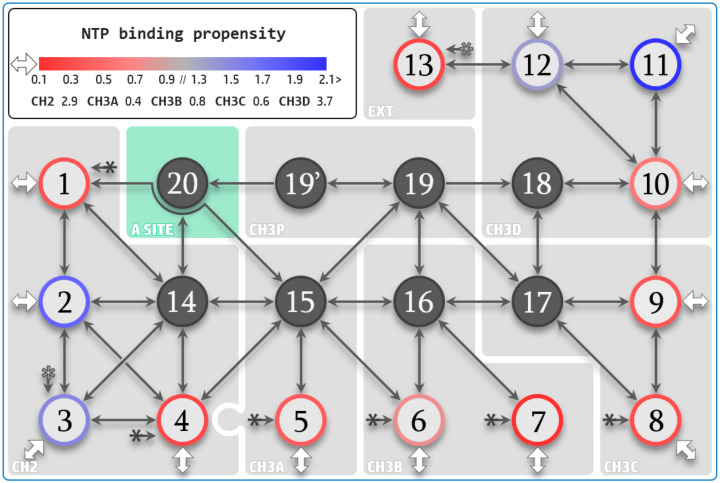

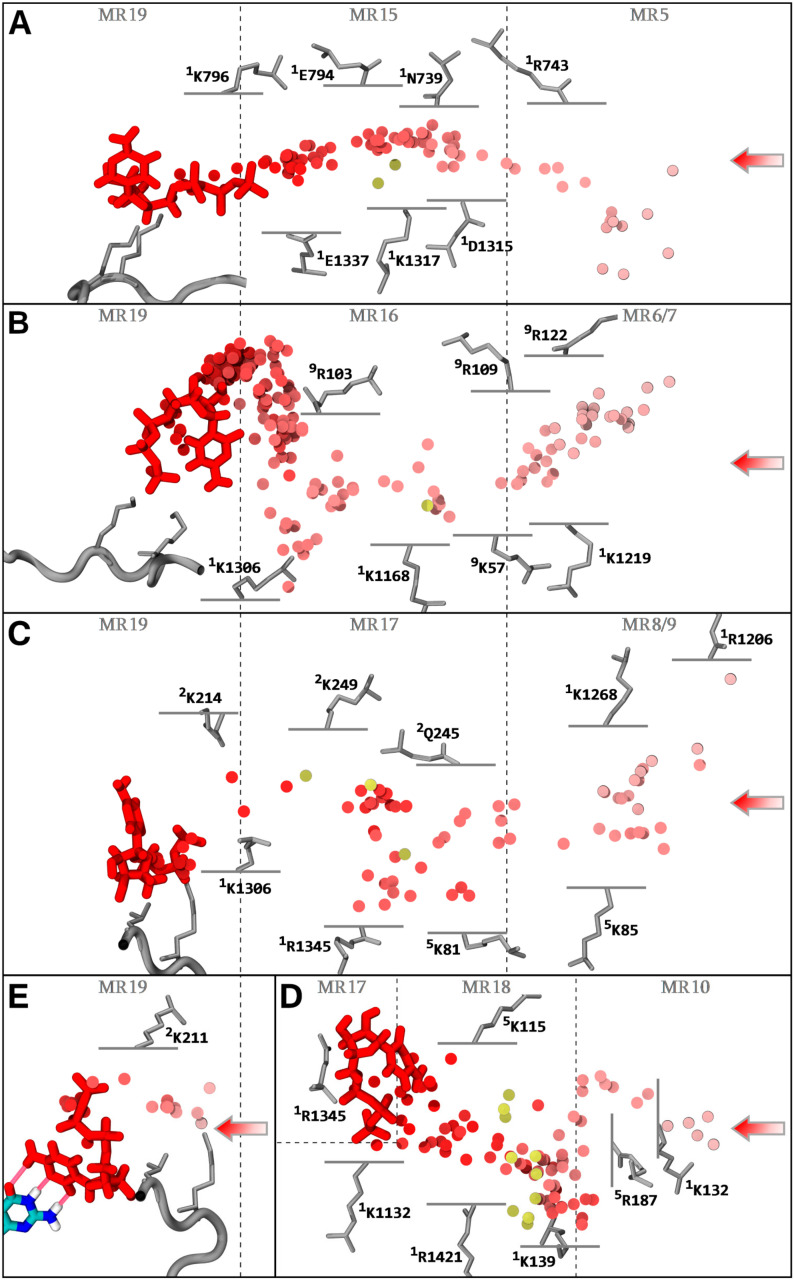

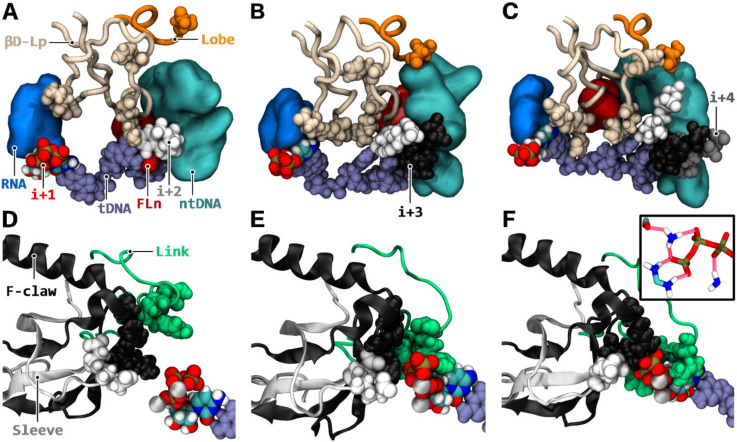
Similar articles
-
Structural Model of RNA Polymerase II Elongation Complex with Complete Transcription Bubble Reveals NTP Entry Routes.PLoS Comput Biol. 2015 Jul 2;11(7):e1004354. doi: 10.1371/journal.pcbi.1004354. eCollection 2015 Jul. PLoS Comput Biol. 2015. PMID: 26134169 Free PMC article.
-
A tunable ratchet driving human RNA polymerase II translocation adjusted by accurately templated nucleoside triphosphates loaded at downstream sites and by elongation factors.J Biol Chem. 2007 Dec 14;282(50):36582-92. doi: 10.1074/jbc.M707014200. Epub 2007 Sep 17. J Biol Chem. 2007. PMID: 17875640
-
Transcription factors IIF and IIS and nucleoside triphosphate substrates as dynamic probes of the human RNA polymerase II mechanism.J Mol Biol. 2004 Sep 24;342(4):1085-99. doi: 10.1016/j.jmb.2004.07.070. J Mol Biol. 2004. PMID: 15351637
-
NTP-driven translocation and regulation of downstream template opening by multi-subunit RNA polymerases.Biochem Cell Biol. 2005 Aug;83(4):486-96. doi: 10.1139/o05-059. Biochem Cell Biol. 2005. PMID: 16094452 Review.
-
DNA bending and wrapping around RNA polymerase: a "revolutionary" model describing transcriptional mechanisms.Microbiol Mol Biol Rev. 1999 Jun;63(2):457-78. doi: 10.1128/MMBR.63.2.457-478.1999. Microbiol Mol Biol Rev. 1999. PMID: 10357858 Free PMC article. Review.
Cited by
-
An NTP-driven mechanism for the nucleotide addition cycle of Escherichia coli RNA polymerase during transcription.PLoS One. 2022 Oct 25;17(10):e0273746. doi: 10.1371/journal.pone.0273746. eCollection 2022. PLoS One. 2022. PMID: 36282801 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
