

Broad-Spectrum Antiviral Entry Inhibition by Interfacially Active Peptides
- PMID: 32907984
- PMCID: PMC7654261
- DOI: 10.1128/JVI.01682-20
Broad-Spectrum Antiviral Entry Inhibition by Interfacially Active Peptides
Abstract
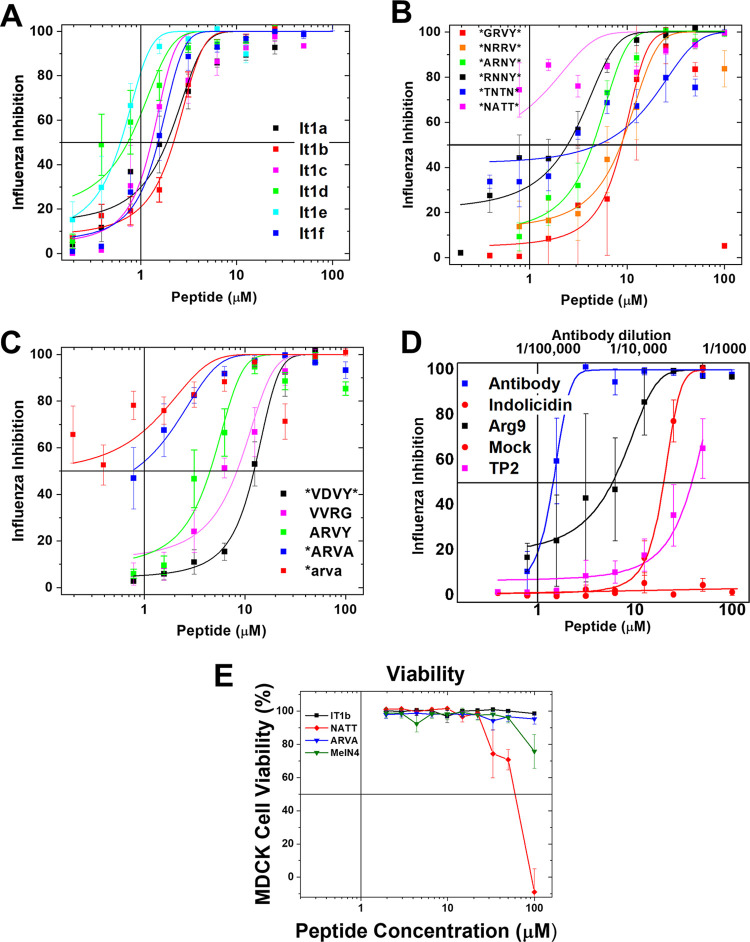
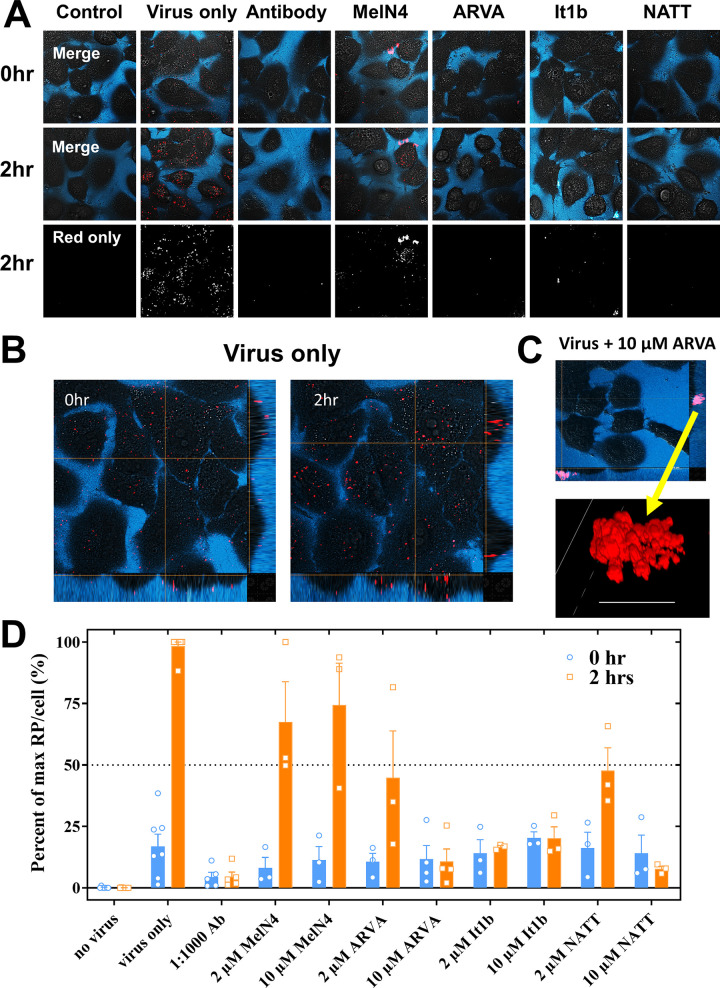
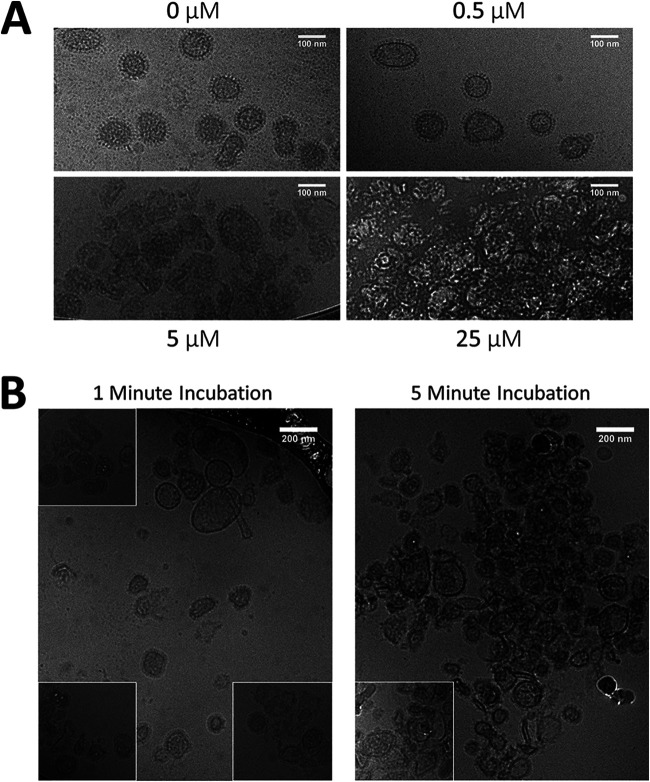
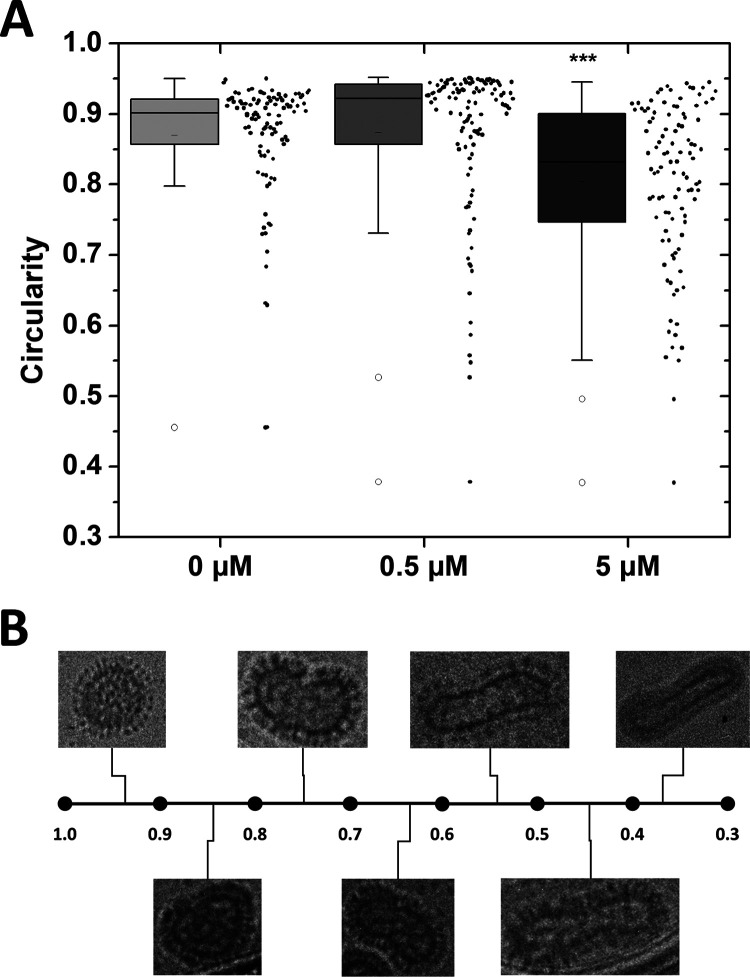
Numerous peptides inhibit the entry of enveloped viruses into cells. Some of these peptides have been shown to inhibit multiple unrelated viruses. We have suggested that such broad-spectrum antiviral peptides share a property called interfacial activity; they are somewhat hydrophobic and amphipathic, with a propensity to interact with the interfacial zones of lipid bilayer membranes. In this study, we further tested the hypothesis that such interfacial activity is a correlate of broad-spectrum antiviral activity. In this study, several families of peptides, selected for the ability to partition into and disrupt membrane integrity but with no known antiviral activity, were tested for the ability to inhibit multiple diverse enveloped viruses. These include Lassa pseudovirus, influenza virus, dengue virus type 2, herpes simplex virus 1, and nonenveloped human adenovirus 5. Various families of interfacially active peptides caused potent inhibition of all enveloped viruses tested at low and submicromolar concentrations, well below the range in which they are toxic to mammalian cells. These membrane-active peptides block uptake and fusion with the host cell by rapidly and directly interacting with virions, destabilizing the viral envelope, and driving virus aggregation and/or intervirion envelope fusion. We speculate that the molecular characteristics shared by these peptides can be exploited to enable the design, optimization, or molecular evolution of novel broad-spectrum antiviral therapeutics.IMPORTANCE New classes of antiviral drugs are needed to treat the ever-changing viral disease landscape. Current antiviral drugs treat only a small number of viral diseases, leaving many patients with established or emerging infections to be treated solely with supportive care. Recent antiviral peptide research has produced numerous membrane-interacting peptides that inhibit diverse enveloped viruses in vitro and in vivo Peptide therapeutics are becoming more common, with over 60 FDA-approved peptides for clinical use. Included in this class of therapeutics is enfuvirtide, a 36-residue peptide drug that inhibits HIV entry/fusion. Due to their broad-spectrum mechanism of action and enormous potential sequence diversity, peptides that inhibit virus entry could potentially fulfill the need for new antiviral therapeutics; however, a better understanding of their mechanism is needed for the optimization or evolution of sequence design to combat the wide landscape of viral disease.
Keywords: entry inhibitor; interfacial activity; membrane; peptide.
Copyright © 2020 American Society for Microbiology.
Figures




References
-
- Heron M. 2019. National Vital Statistics Reports deaths: leading causes for 2017. Natl Vital Stat Rep 68:1–77. - PubMed
-
- Murphy SL, Xu J, Kochanek KD, Arias E. 2018. Mortality in the United States, 2017. NCHS Data Brief 2018(328):1–8. - PubMed
-
- Johnson NB, Hayes LD, Brown K, Hoo EC, Ethier KA, Centers for Disease Control and Prevention. 2014. CDC National Health Report: leading causes of morbidity and mortality and associated behavioral risk and protective factors—United States, 2005–2013. MMWR Suppl 63:3–27. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
