

An intact gut microbiome protects genetically predisposed mice against leukemia
- PMID: 32911536
- PMCID: PMC7694022
- DOI: 10.1182/blood.2019004381
An intact gut microbiome protects genetically predisposed mice against leukemia
Abstract
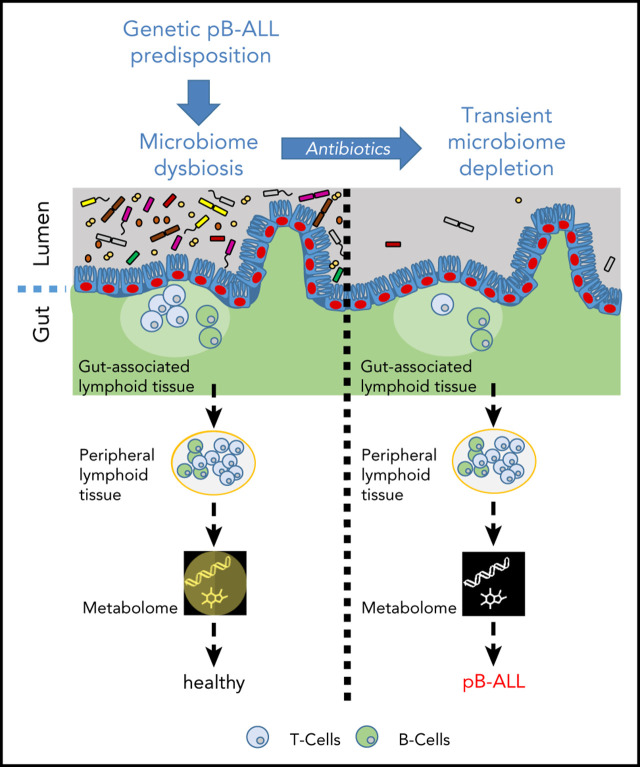
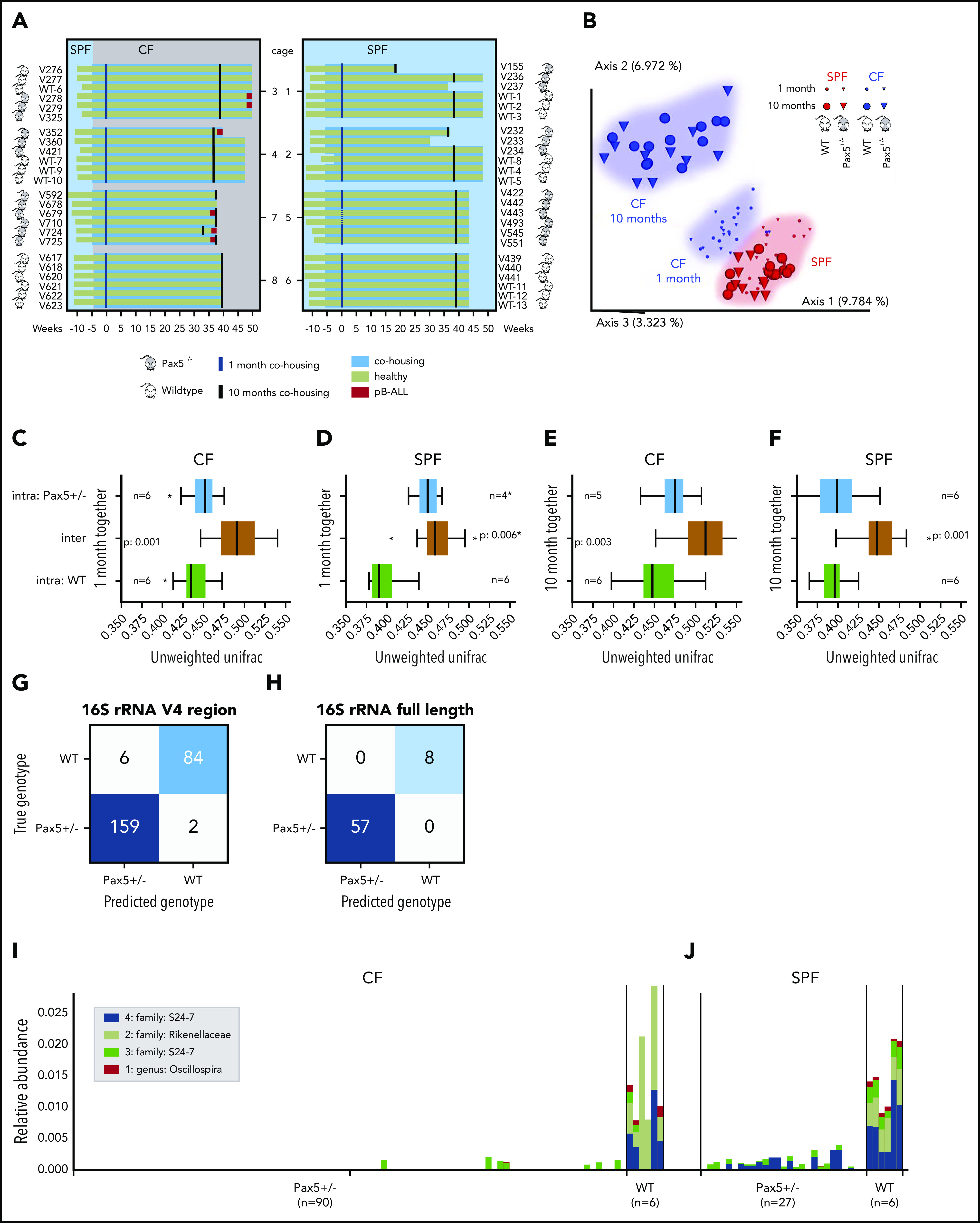
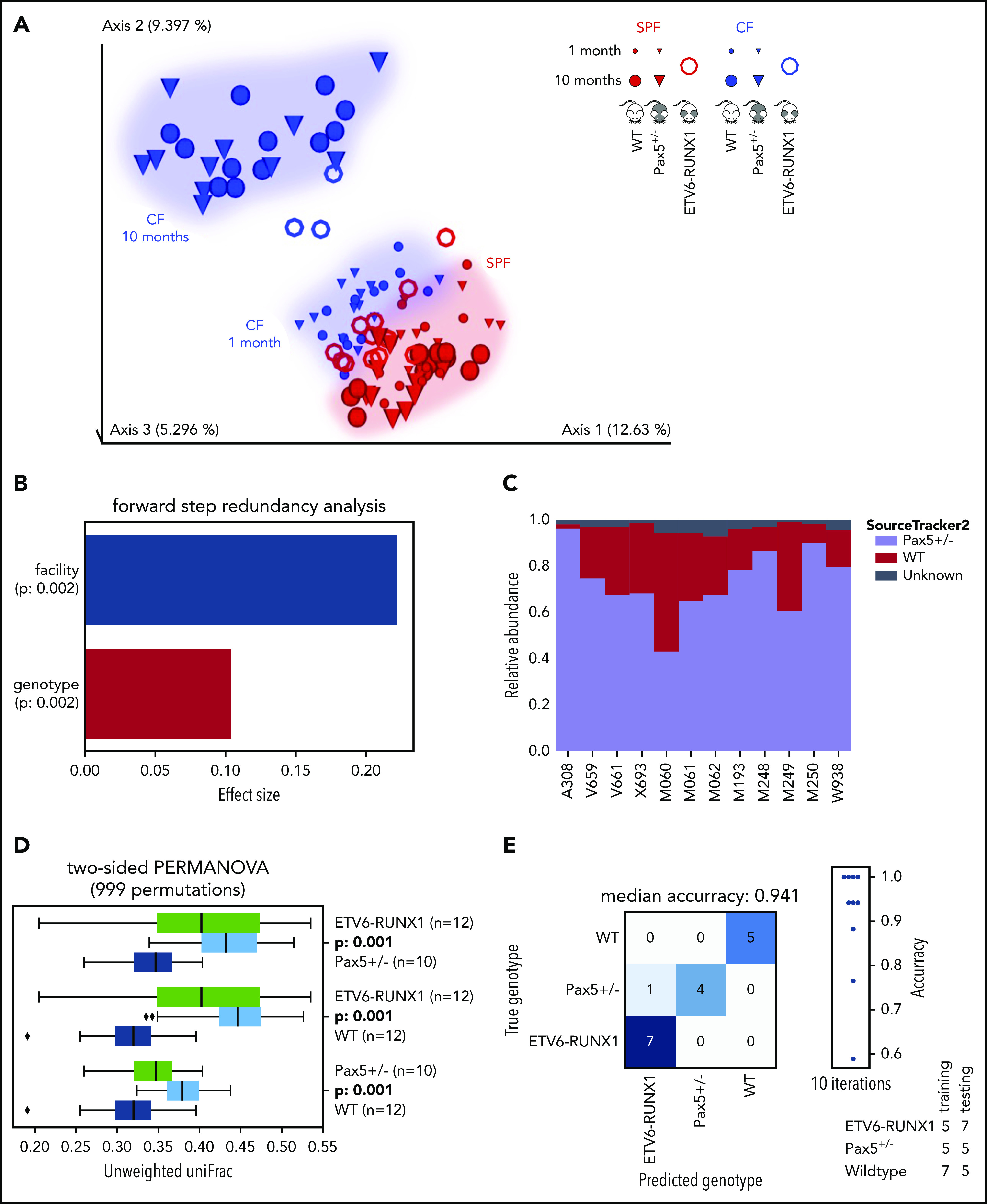
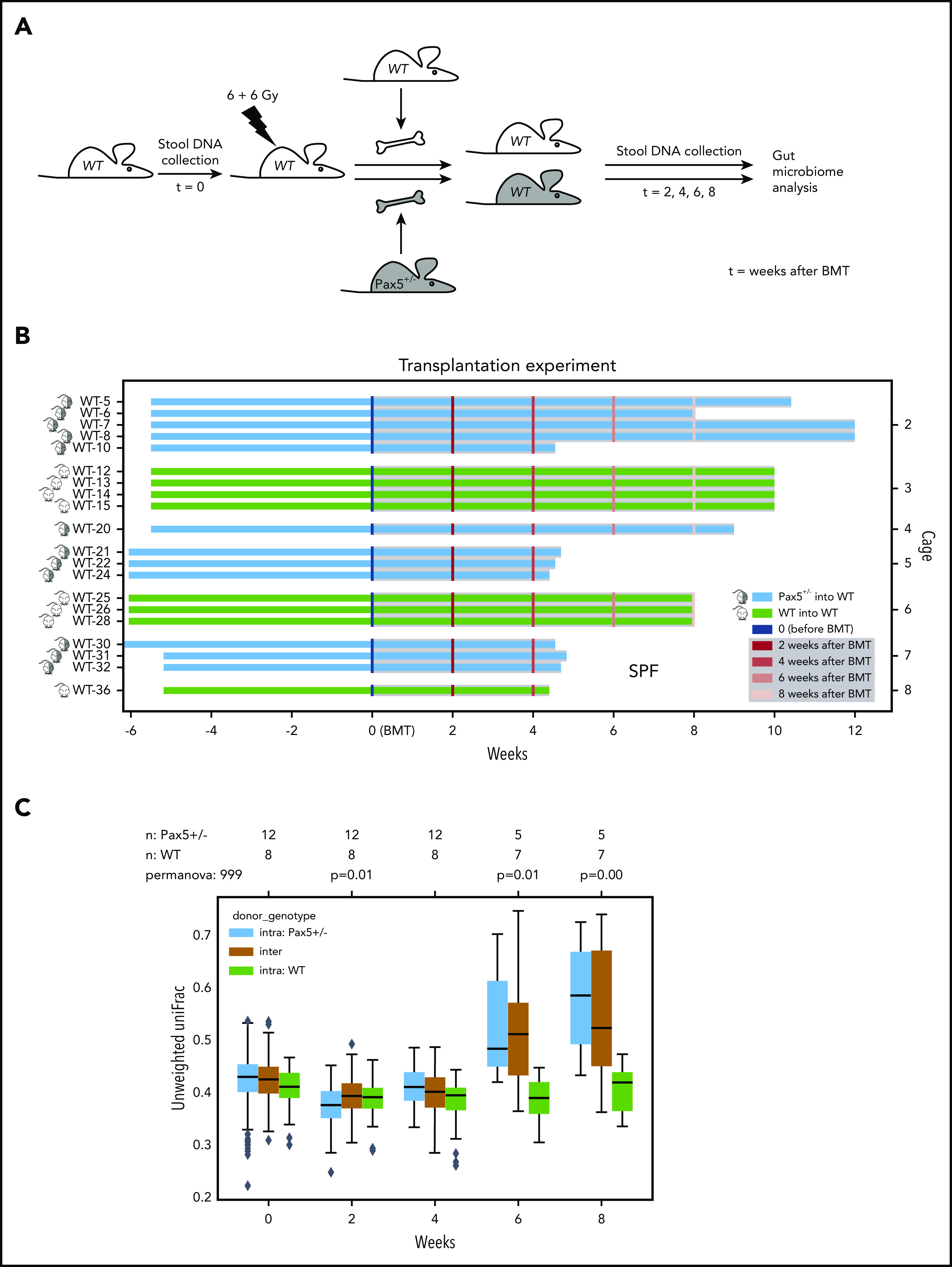
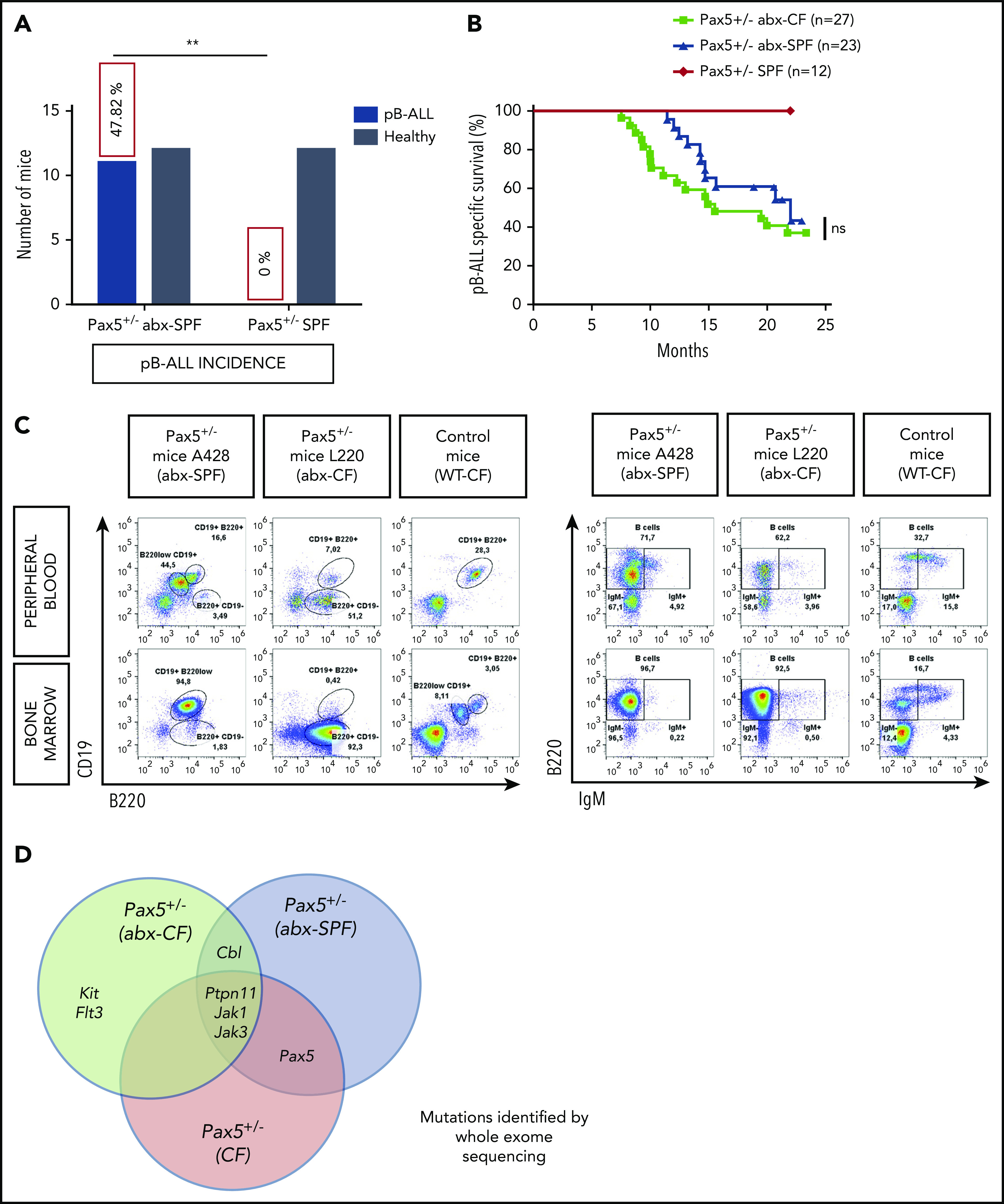
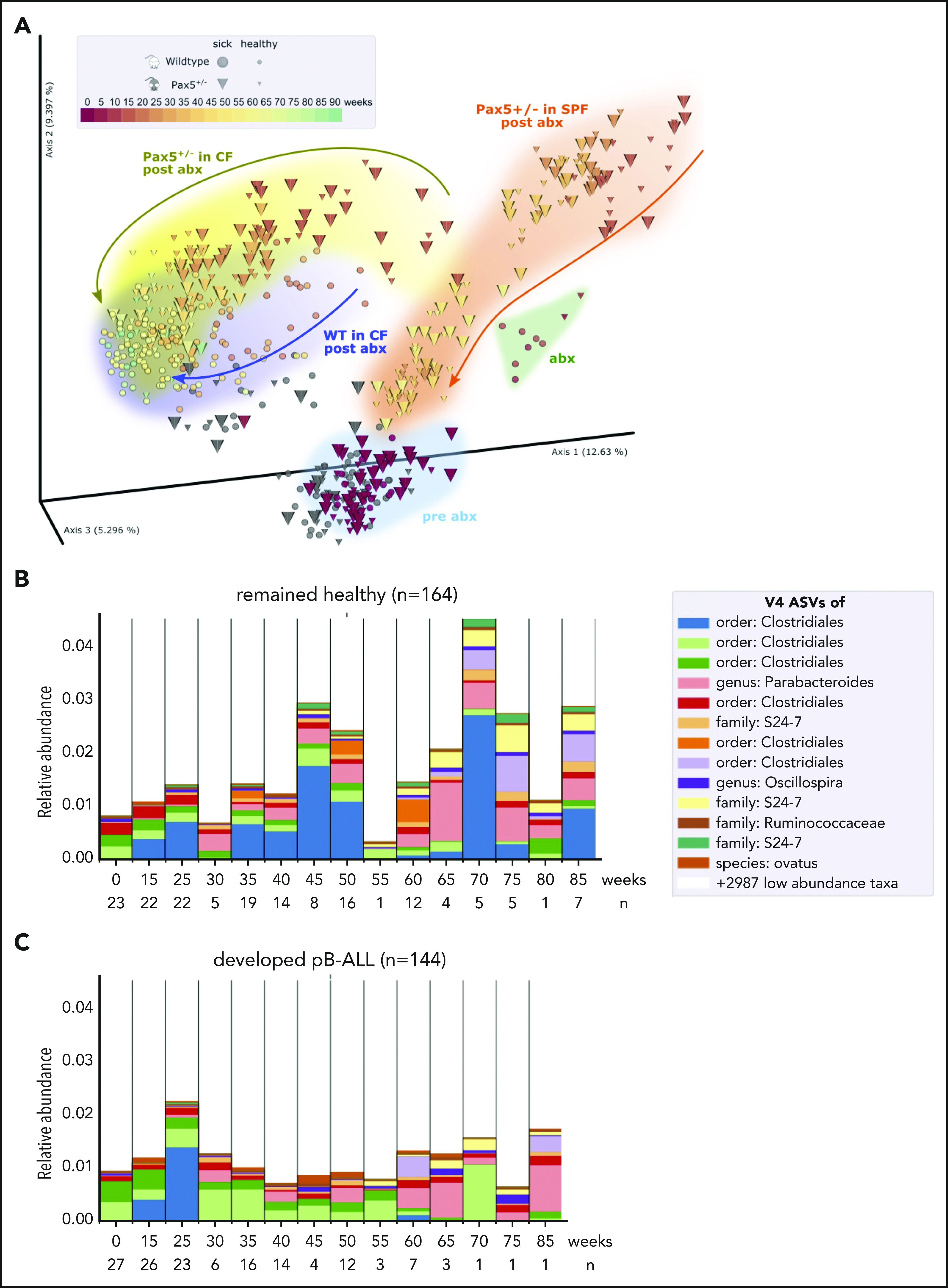
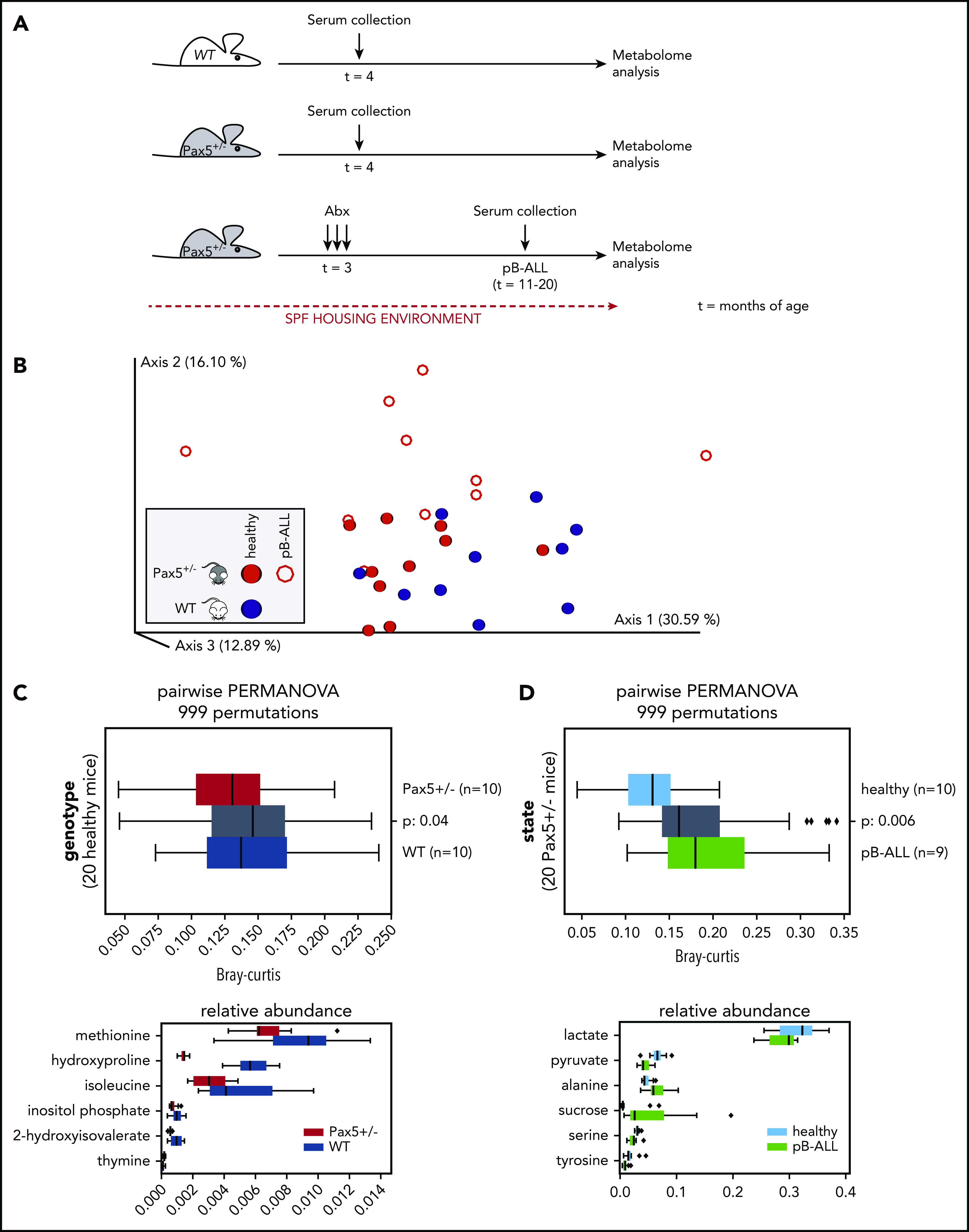
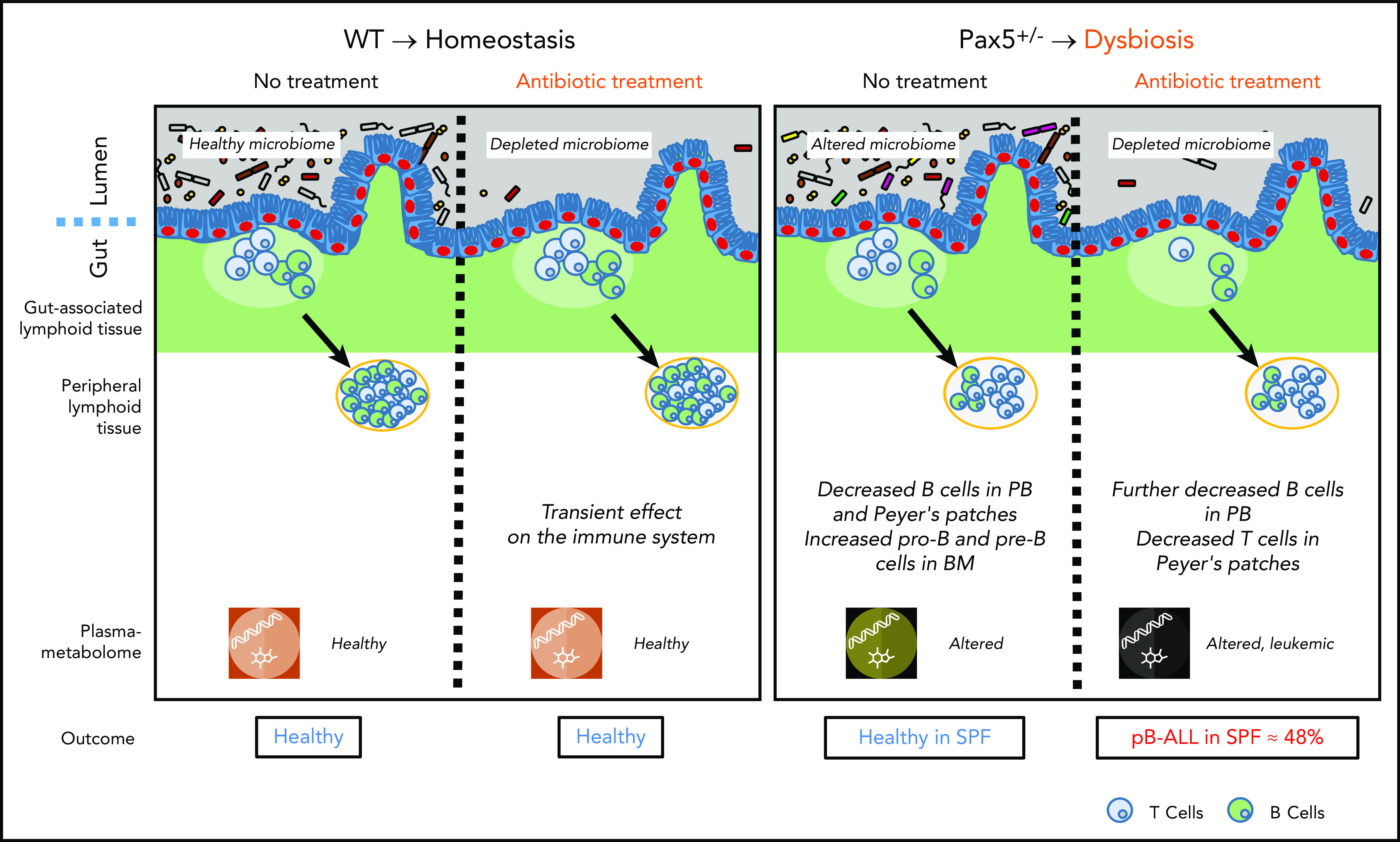
The majority of childhood leukemias are precursor B-cell acute lymphoblastic leukemias (pB-ALLs) caused by a combination of prenatal genetic predispositions and oncogenic events occurring after birth. Although genetic predispositions are frequent in children (>1% to 5%), fewer than 1% of genetically predisposed carriers will develop pB-ALL. Although infectious stimuli are believed to play a major role in leukemogenesis, the critical determinants are not well defined. Here, by using murine models of pB-ALL, we show that microbiome disturbances incurred by antibiotic treatment early in life were sufficient to induce leukemia in genetically predisposed mice, even in the absence of infectious stimuli and independent of T cells. By using V4 and full-length 16S ribosomal RNA sequencing of a series of fecal samples, we found that genetic predisposition to pB-ALL (Pax5 heterozygosity or ETV6-RUNX1 fusion) shaped a distinct gut microbiome. Machine learning accurately (96.8%) predicted genetic predisposition using 40 of 3983 amplicon sequence variants as proxies for bacterial species. Transplantation of either wild-type (WT) or Pax5+/- hematopoietic bone marrow cells into WT recipient mice revealed that the microbiome is shaped and determined in a donor genotype-specific manner. Gas chromatography-mass spectrometry (GC-MS) analyses of sera from WT and Pax5+/- mice demonstrated the presence of a genotype-specific distinct metabolomic profile. Taken together, our data indicate that it is a lack of commensal microbiota rather than the presence of specific bacteria that promotes leukemia in genetically predisposed mice. Future large-scale longitudinal studies are required to determine whether targeted microbiome modification in children predisposed to pB-ALL could become a successful prevention strategy.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
Comment in
-
A "gut feeling" about precursor B-ALL.Blood. 2020 Oct 29;136(18):1995-1996. doi: 10.1182/blood.2020008907. Blood. 2020. PMID: 33119762 No abstract available.
References
-
- Parkin DM, Stiller CA, Draper GJ, Bieber CA. The international incidence of childhood cancer. Int J Cancer. 1988;42(4):511-520. - PubMed
-
- Linabery AM, Ross JA. Trends in childhood cancer incidence in the U.S. (1992-2004). Cancer. 2008;112(2):416-432. - PubMed
-
- Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematology Am Soc Hematol Educ Program. 2014;2014(1):174-180. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
