

Two Novel Pathogenic Variants Confirm RMND1 Causative Role in Perrault Syndrome with Renal Involvement
- PMID: 32911714
- PMCID: PMC7564844
- DOI: 10.3390/genes11091060
Two Novel Pathogenic Variants Confirm RMND1 Causative Role in Perrault Syndrome with Renal Involvement
Abstract
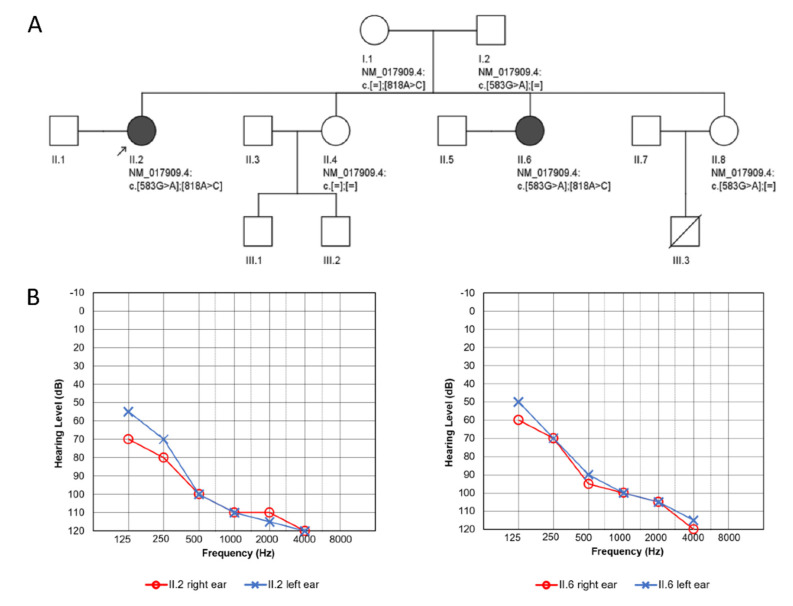
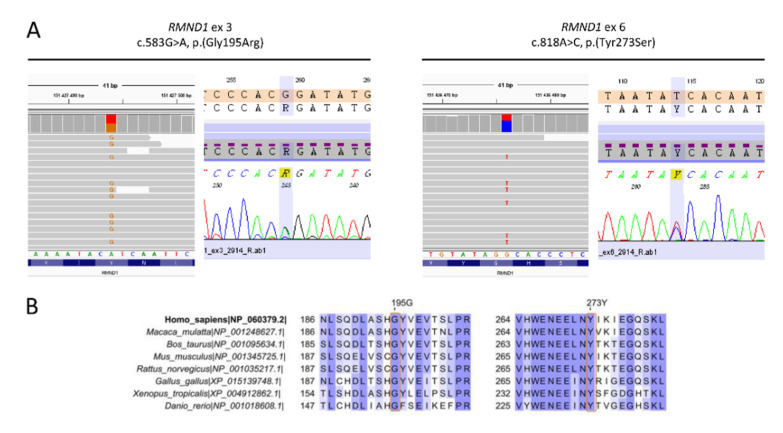
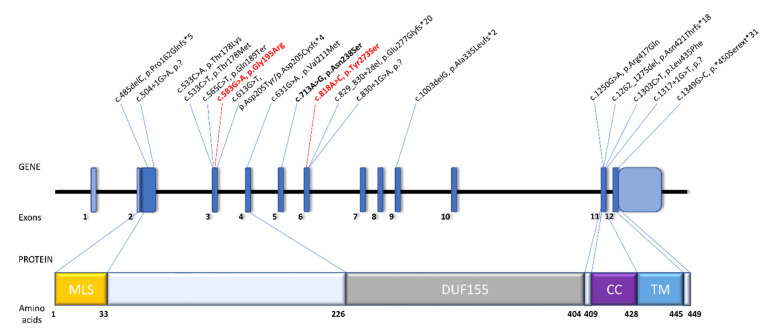
RMND1 (required for meiotic nuclear division 1 homolog) pathogenic variants are known to cause combined oxidative phosphorylation deficiency (COXPD11), a severe multisystem disorder. In one patient, a homozygous RMND1 pathogenic variant, with an established role in COXPD11, was associated with a Perrault-like syndrome. We performed a thorough clinical investigation and applied a targeted multigene hearing loss panel to reveal the cause of hearing loss, ovarian dysfunction (two cardinal features of Perrault syndrome) and chronic kidney disease in two adult female siblings. Two compound heterozygous missense variants, c.583G>A (p.Gly195Arg) and c.818A>C (p.Tyr273Ser), not previously associated with disease, were identified in RMND1 in both patients, and their segregation with disease was confirmed in family members. The patients have no neurological or intellectual impairment, and nephrological evaluation predicts a benign course of kidney disease. Our study presents the mildest, so far reported, RMND1-related phenotype and delivers the first independent confirmation that RMND1 is causally involved in the development of Perrault syndrome with renal involvement. This highlights the importance of including RMND1 to the list of Perrault syndrome causative factors and provides new insight into the clinical manifestation of RMND1 deficiency.
Keywords: COXPD11 (combined oxidative phosphorylation deficiency); Perrault syndrome; RMND1 (required for meiotic nuclear division 1 homolog); hearing loss; mitochondria; ovarian dysfunction; renal disease.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Garcia-Diaz B., Barros M.H., Sanna-Cherchi S., Emmanuele V., Akman H.O., Ferreiro-Barros C.C., Horvath R., Tadesse S., El Gharaby N., DiMauro S., et al. Infantile encephaloneuromyopathy and defective mitochondrial translation are due to a homozygous RMND1 mutation. Am. J. Hum. Genet. 2012;91:729–736. doi: 10.1016/j.ajhg.2012.08.019. - DOI - PMC - PubMed
-
- Janer A., Antonicka H., Lalonde E., Nishimura T., Sasarman F., Brown G.K., Brown R.M., Majewski J., Shoubridge E.A. An RMND1 Mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am. J. Hum. Genet. 2012;91:737–743. doi: 10.1016/j.ajhg.2012.08.020. - DOI - PMC - PubMed
-
- Janer A., van Karnebeek C.D., Sasarman F., Antonicka H., Al Ghamdi M., Shyr C., Dunbar M., Stockler-Ispiroglu S., Ross C.J., Vallance H., et al. RMND1 deficiency associated with neonatal lactic acidosis, infantile onset renal failure, deafness, and multiorgan involvement. Eur. J. Hum. Genet. 2015;23:1301–1307. doi: 10.1038/ejhg.2014.293. - DOI - PMC - PubMed
-
- Ng Y.S., Alston C.L., Diodato D., Morris A.A., Ulrick N., Kmoch S., Houstek J., Martinelli D., Haghighi A., Atiq M., et al. The clinical, biochemical and genetic features associated with RMND1-related mitochondrial disease. J. Med. Genet. 2016;53:768–775. doi: 10.1136/jmedgenet-2016-103910. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
