

Transcriptomic signatures across human tissues identify functional rare genetic variation
- PMID: 32913073
- PMCID: PMC7646251
- DOI: 10.1126/science.aaz5900
Transcriptomic signatures across human tissues identify functional rare genetic variation
Abstract
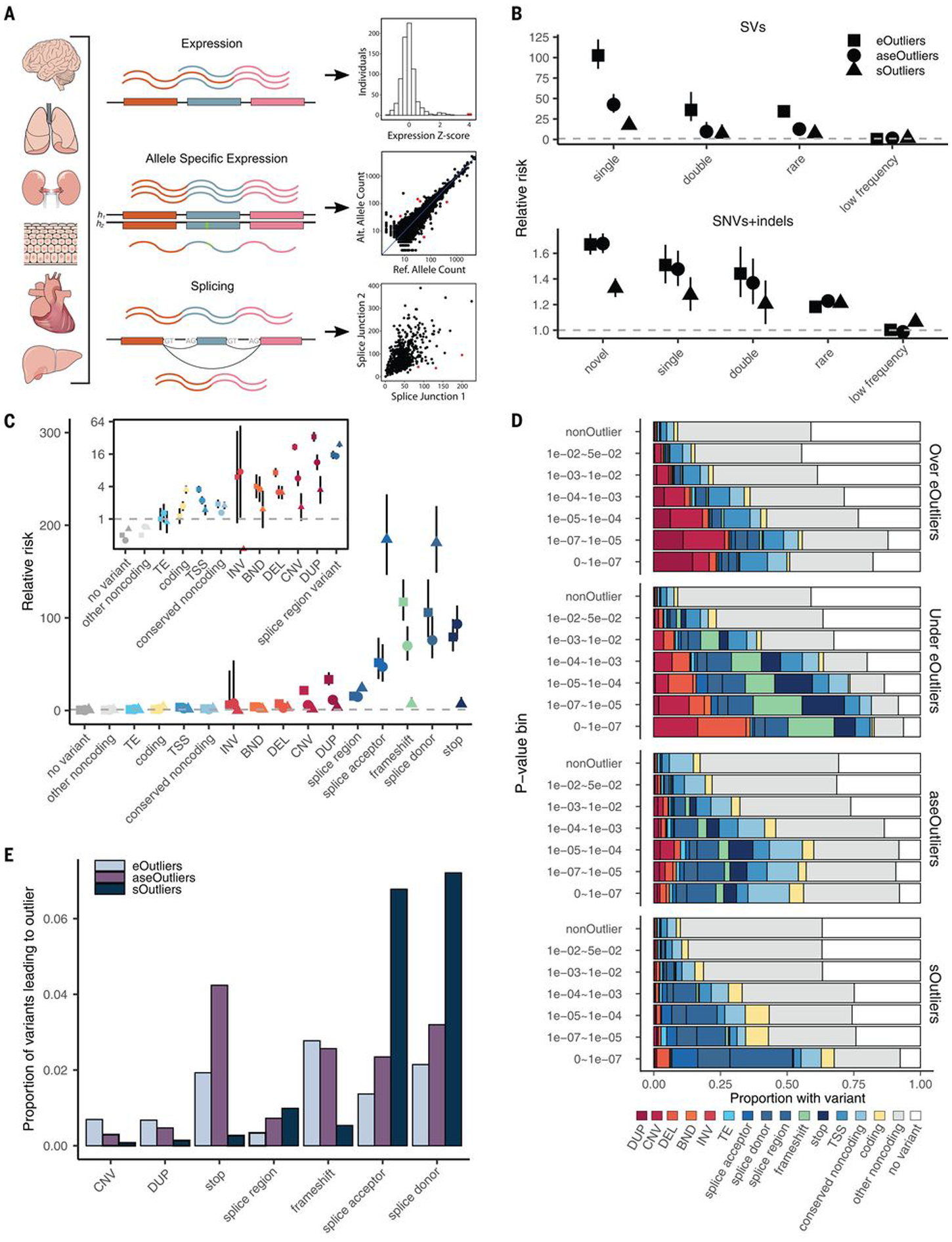
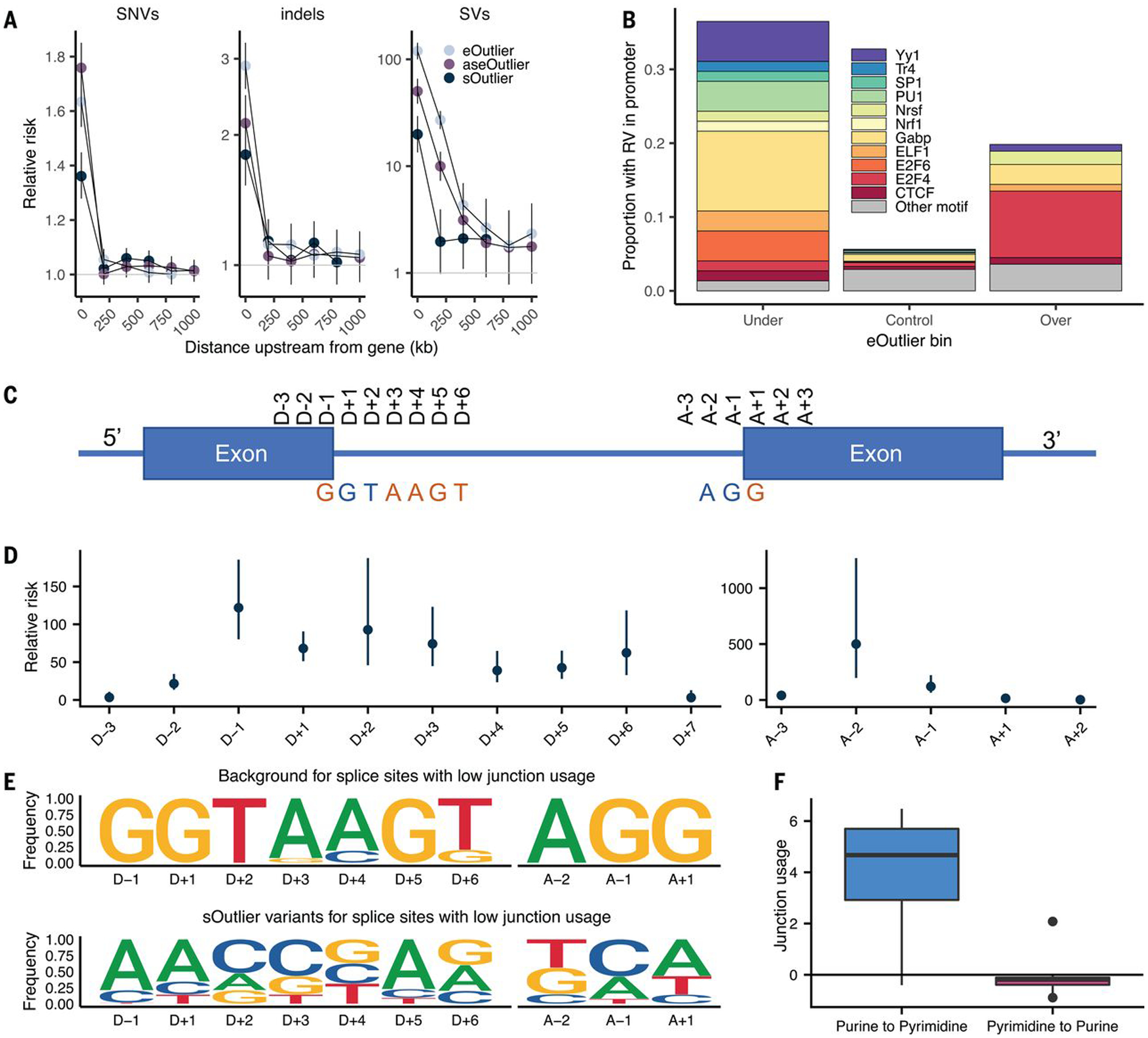
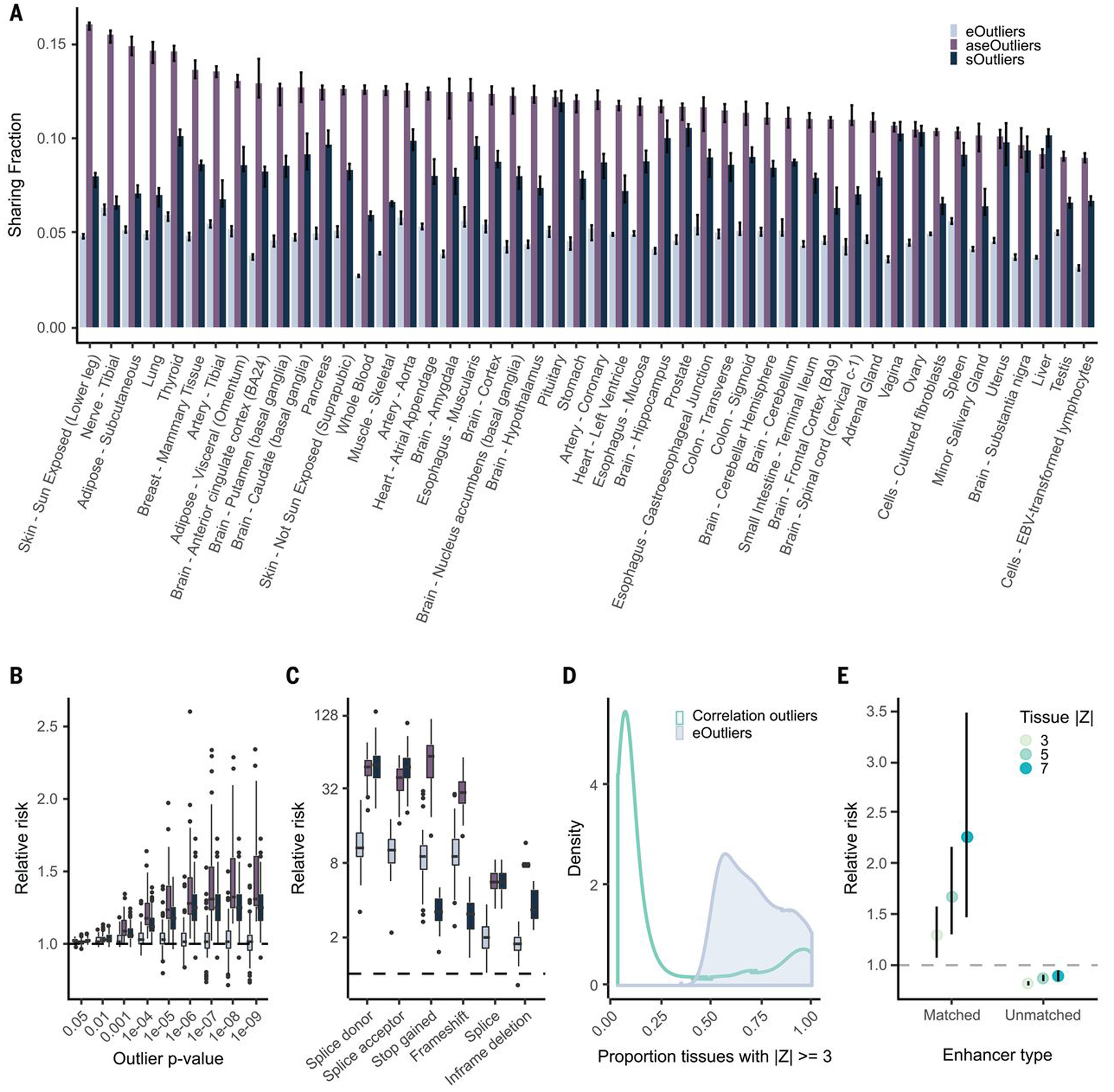
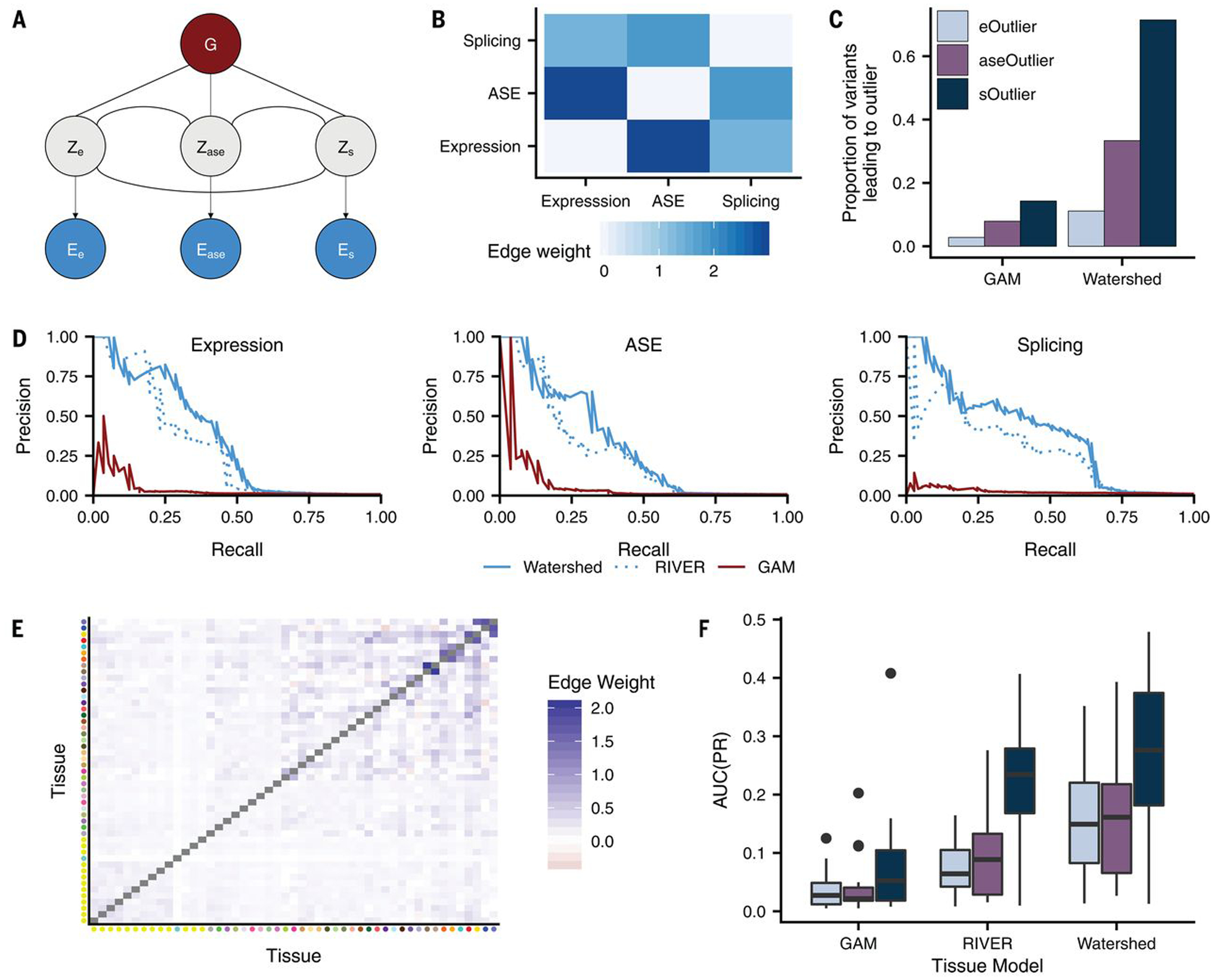
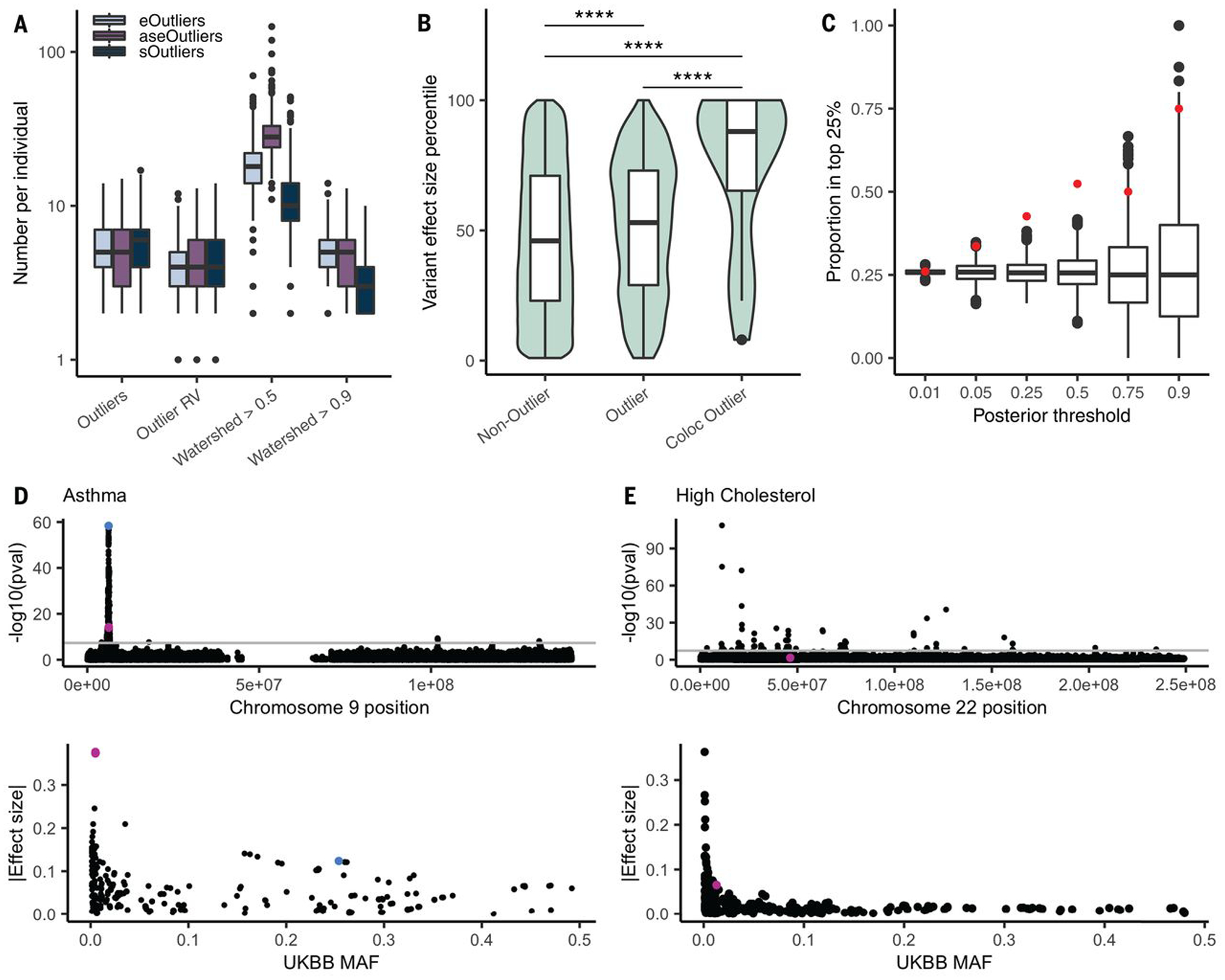
Rare genetic variants are abundant across the human genome, and identifying their function and phenotypic impact is a major challenge. Measuring aberrant gene expression has aided in identifying functional, large-effect rare variants (RVs). Here, we expanded detection of genetically driven transcriptome abnormalities by analyzing gene expression, allele-specific expression, and alternative splicing from multitissue RNA-sequencing data, and demonstrate that each signal informs unique classes of RVs. We developed Watershed, a probabilistic model that integrates multiple genomic and transcriptomic signals to predict variant function, validated these predictions in additional cohorts and through experimental assays, and used them to assess RVs in the UK Biobank, the Million Veterans Program, and the Jackson Heart Study. Our results link thousands of RVs to diverse molecular effects and provide evidence to associate RVs affecting the transcriptome with human traits.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
Comment in
-
Reaching completion for GTEx.Nat Rev Genet. 2020 Dec;21(12):717. doi: 10.1038/s41576-020-00296-7. Nat Rev Genet. 2020. PMID: 33060849 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- HHSN268201100037C/HL/NHLBI NIH HHS/United States
- HHSN268201800012C/HL/NHLBI NIH HHS/United States
- R01 HL120393/HL/NHLBI NIH HHS/United States
- R01 MH106842/MH/NIMH NIH HHS/United States
- HHSN268201800014C/HL/NHLBI NIH HHS/United States
- R01 HG006855/HG/NHGRI NIH HHS/United States
- T32 HG000044/HG/NHGRI NIH HHS/United States
- HHSN268201800011I/HB/NHLBI NIH HHS/United States
- T32 LM012409/LM/NLM NIH HHS/United States
- T32 GM007057/GM/NIGMS NIH HHS/United States
- U01 HG009431/HG/NHGRI NIH HHS/United States
- U01 MH104393/MH/NIMH NIH HHS/United States
- R01 HG010480/HG/NHGRI NIH HHS/United States
- R01 DA006227/DA/NIDA NIH HHS/United States
- U41 HG002371/HG/NHGRI NIH HHS/United States
- R01 HG008150/HG/NHGRI NIH HHS/United States
- HHSN268201800014I/HB/NHLBI NIH HHS/United States
- UL1 TR001114/TR/NCATS NIH HHS/United States
- R01 MH107666/MH/NIMH NIH HHS/United States
- U01 HL120393/HL/NHLBI NIH HHS/United States
- R01 MH109905/MH/NIMH NIH HHS/United States
- R01 HL077612/HL/NHLBI NIH HHS/United States
- UM1 HG008901/HG/NHGRI NIH HHS/United States
- U01 HG007593/HG/NHGRI NIH HHS/United States
- R01 GM124486/GM/NIGMS NIH HHS/United States
- R01 MH090936/MH/NIMH NIH HHS/United States
- U01 HG007598/HG/NHGRI NIH HHS/United States
- R01 HL142711/HL/NHLBI NIH HHS/United States
- R01 HG009125/HG/NHGRI NIH HHS/United States
- T32 DK110919/DK/NIDDK NIH HHS/United States
- R01 HG002585/HG/NHGRI NIH HHS/United States
- HHSN268201800001C/HL/NHLBI NIH HHS/United States
- R01 HL142015/HL/NHLBI NIH HHS/United States
- HHSN268201800013I/MD/NIMHD NIH HHS/United States
- K99 HG009916/HG/NHGRI NIH HHS/United States
- T15 LM007033/LM/NLM NIH HHS/United States
- R01 HL142028/HL/NHLBI NIH HHS/United States
- I01 BX003340/BX/BLRD VA/United States
- HHSN268201800012I/HL/NHLBI NIH HHS/United States
- U01 HG009080/HG/NHGRI NIH HHS/United States
- HHSN268201800011C/HL/NHLBI NIH HHS/United States
- R01 MH090951/MH/NIMH NIH HHS/United States
- I01 BX003362/BX/BLRD VA/United States
- UL1 TR002550/TR/NCATS NIH HHS/United States
- F32 HG009987/HG/NHGRI NIH HHS/United States
- R01 HG010067/HG/NHGRI NIH HHS/United States
- HHSN268201800015I/HB/NHLBI NIH HHS/United States
- R01 MH090941/MH/NIMH NIH HHS/United States
- R01 AG066490/AG/NIA NIH HHS/United States
- R01 GM122924/GM/NIGMS NIH HHS/United States
- U41 HG009494/HG/NHGRI NIH HHS/United States
- HHSN268201800010I/HB/NHLBI NIH HHS/United States
- R01 MH101822/MH/NIMH NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- UL1 TR001873/TR/NCATS NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
- R35 HG010718/HG/NHGRI NIH HHS/United States
- P30 DK020595/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
