

Efficient low-cost chromatin profiling with CUT&Tag
- PMID: 32913232
- PMCID: PMC8318778
- DOI: 10.1038/s41596-020-0373-x
Efficient low-cost chromatin profiling with CUT&Tag
Abstract
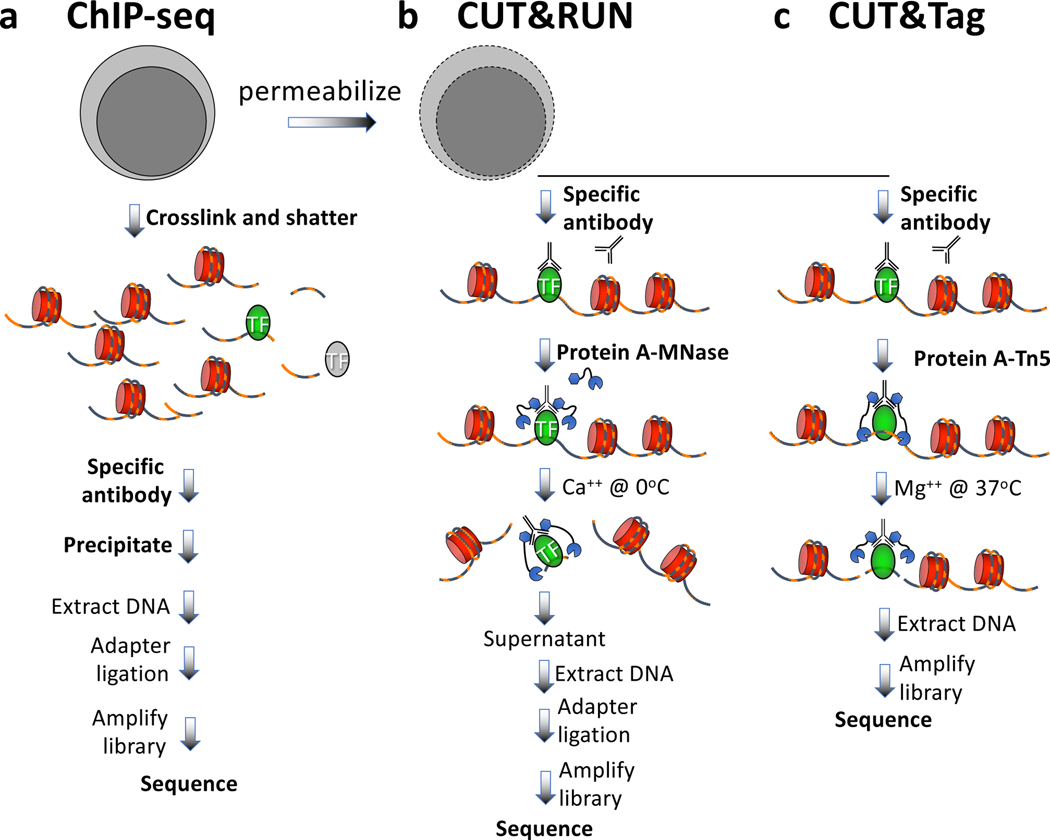
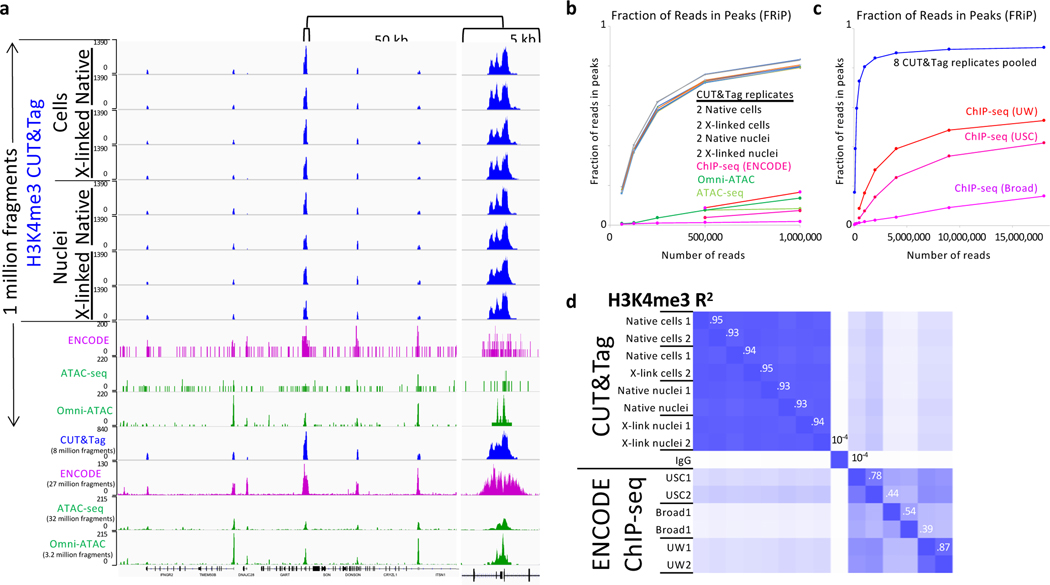
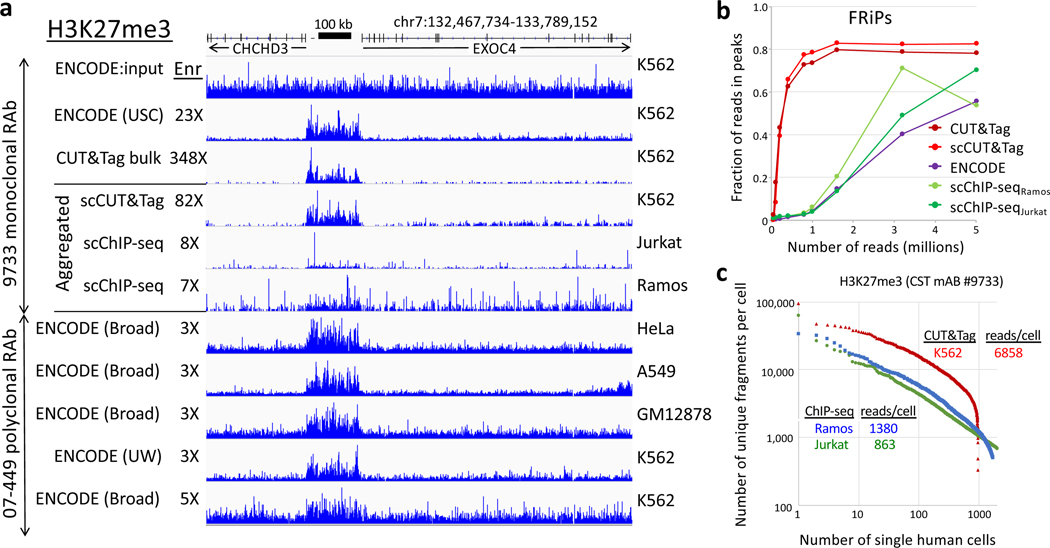
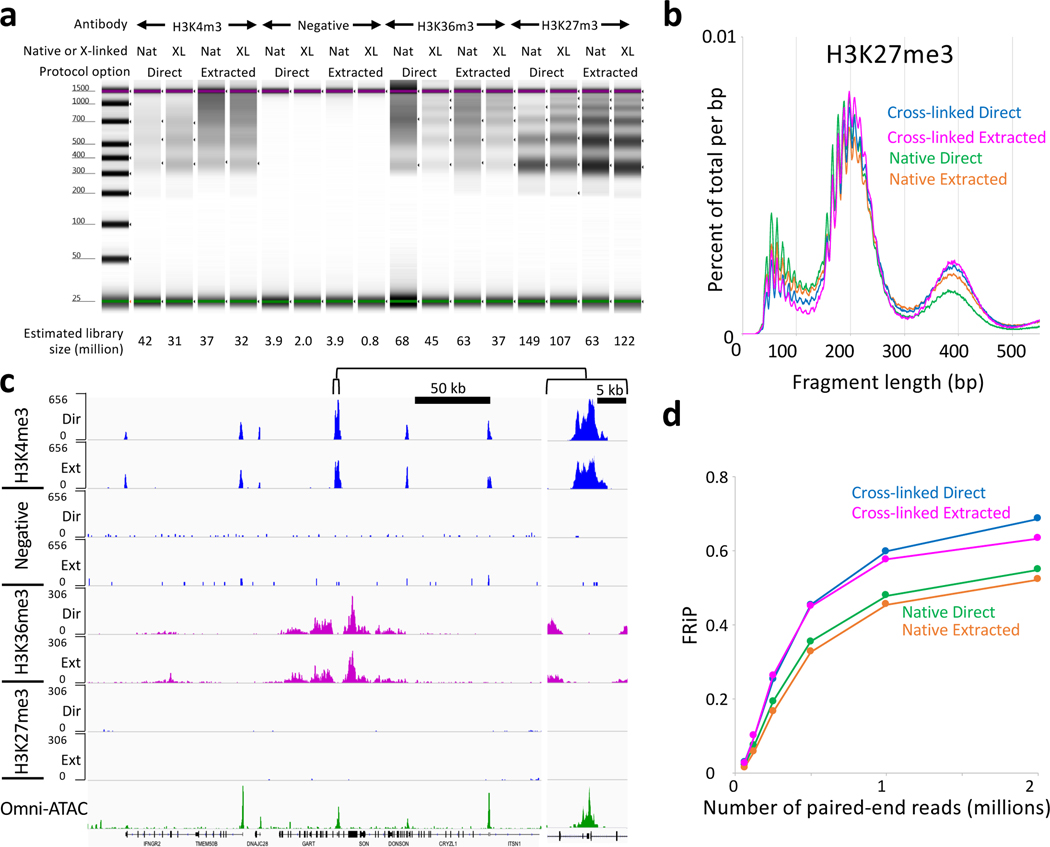
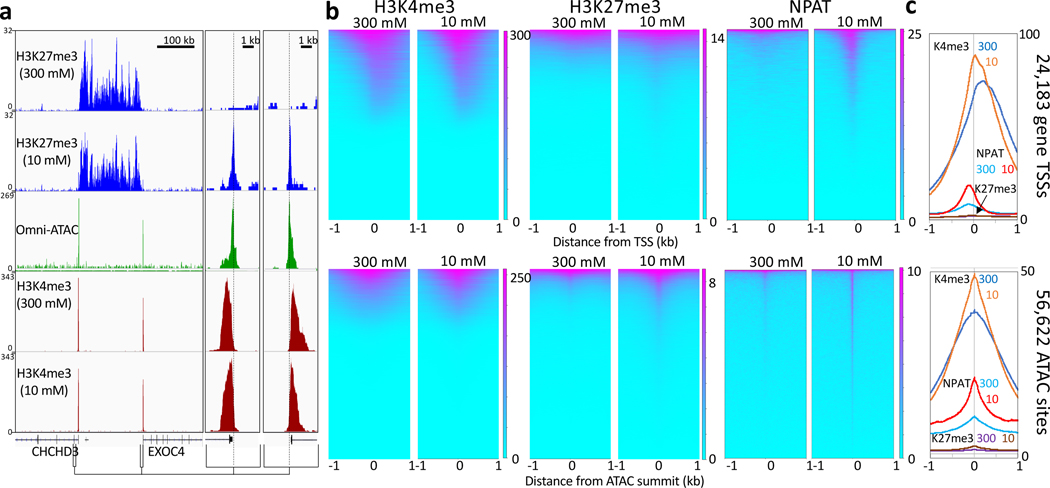
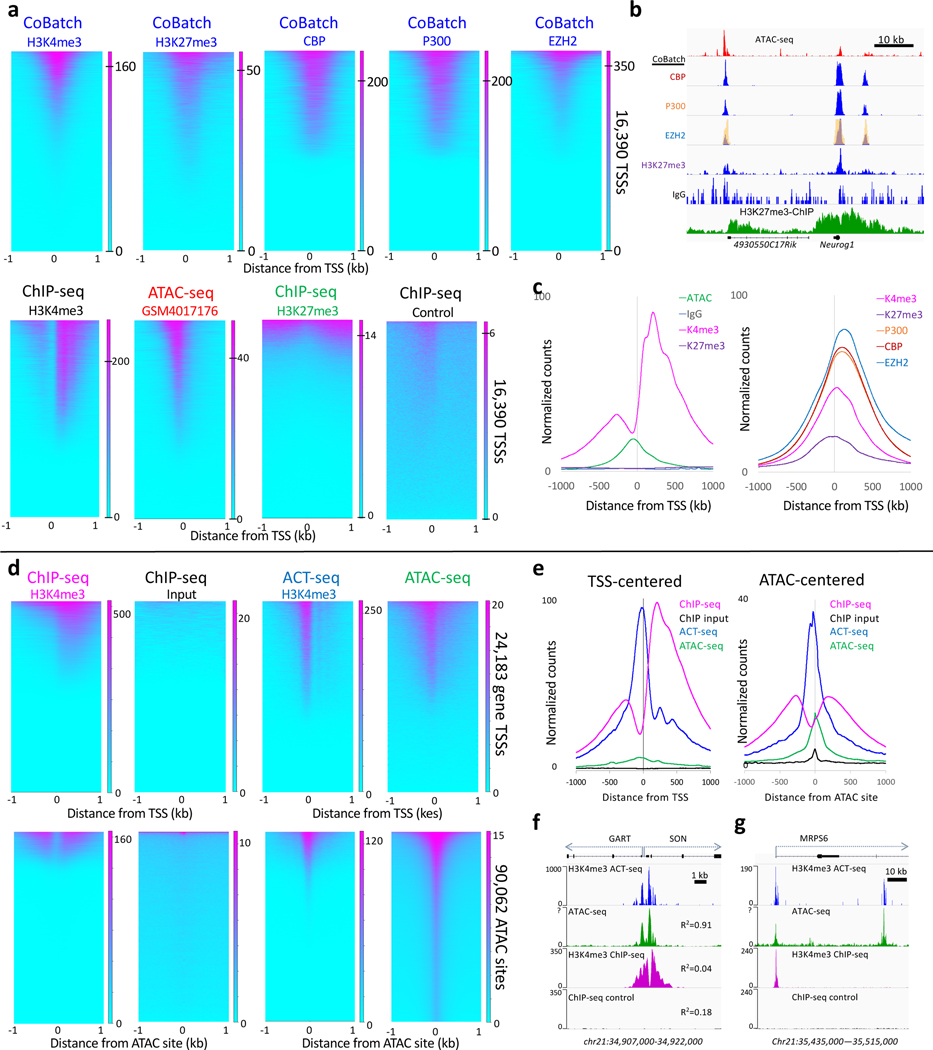
We recently introduced Cleavage Under Targets & Tagmentation (CUT&Tag), an epigenomic profiling strategy in which antibodies are bound to chromatin proteins in situ in permeabilized nuclei. These antibodies are then used to tether the cut-and-paste transposase Tn5. Activation of the transposase simultaneously cleaves DNA and adds adapters ('tagmentation') for paired-end DNA sequencing. Here, we introduce a streamlined CUT&Tag protocol that suppresses DNA accessibility artefacts to ensure high-fidelity mapping of the antibody-targeted protein and improves the signal-to-noise ratio over current chromatin profiling methods. Streamlined CUT&Tag can be performed in a single PCR tube, from cells to amplified libraries, providing low-cost genome-wide chromatin maps. By simplifying library preparation CUT&Tag requires less than a day at the bench, from live cells to sequencing-ready barcoded libraries. As a result of low background levels, barcoded and pooled CUT&Tag libraries can be sequenced for as little as $25 per sample. This enables routine genome-wide profiling of chromatin proteins and modifications and requires no special skills or equipment.
Figures
References
-
- Rodriguez-Ubreva J & Ballestar E Chromatin immunoprecipitation. Methods Mol Biol 1094, 309–18 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
