

Amino acid depletion triggered by ʟ-asparaginase sensitizes MM cells to carfilzomib by inducing mitochondria ROS-mediated cell death
- PMID: 32915979
- PMCID: PMC7509874
- DOI: 10.1182/bloodadvances.2020001639
Amino acid depletion triggered by ʟ-asparaginase sensitizes MM cells to carfilzomib by inducing mitochondria ROS-mediated cell death
Abstract
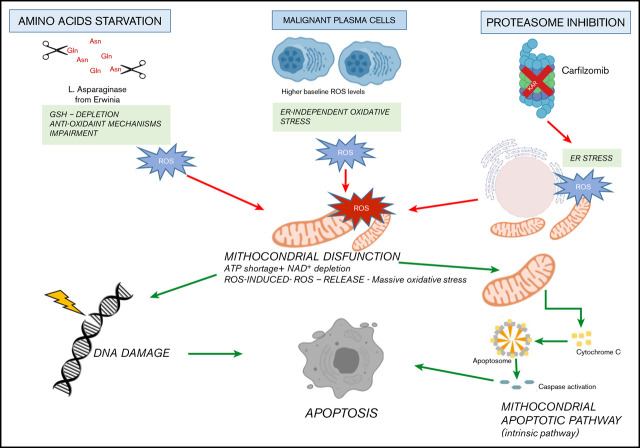
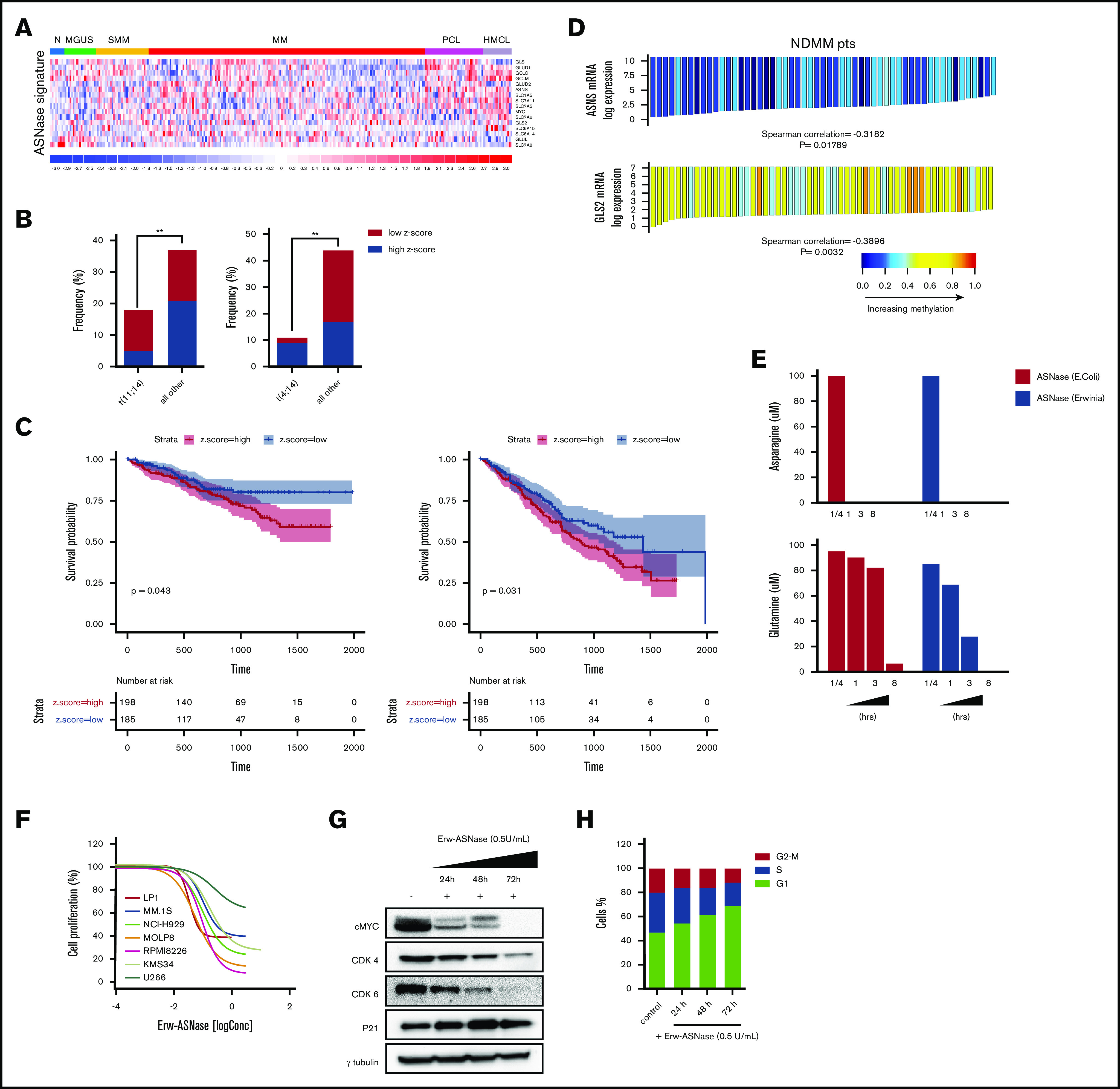
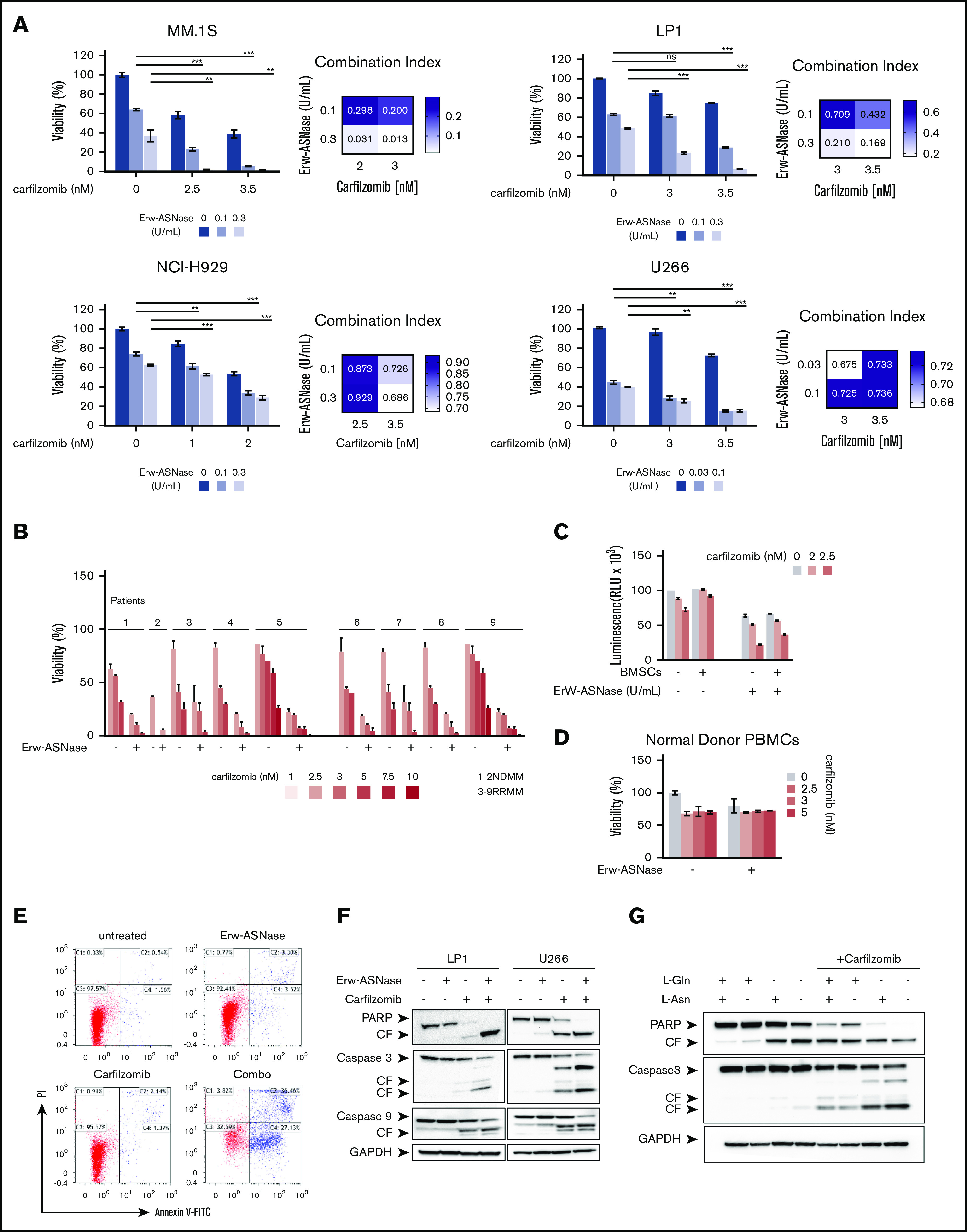
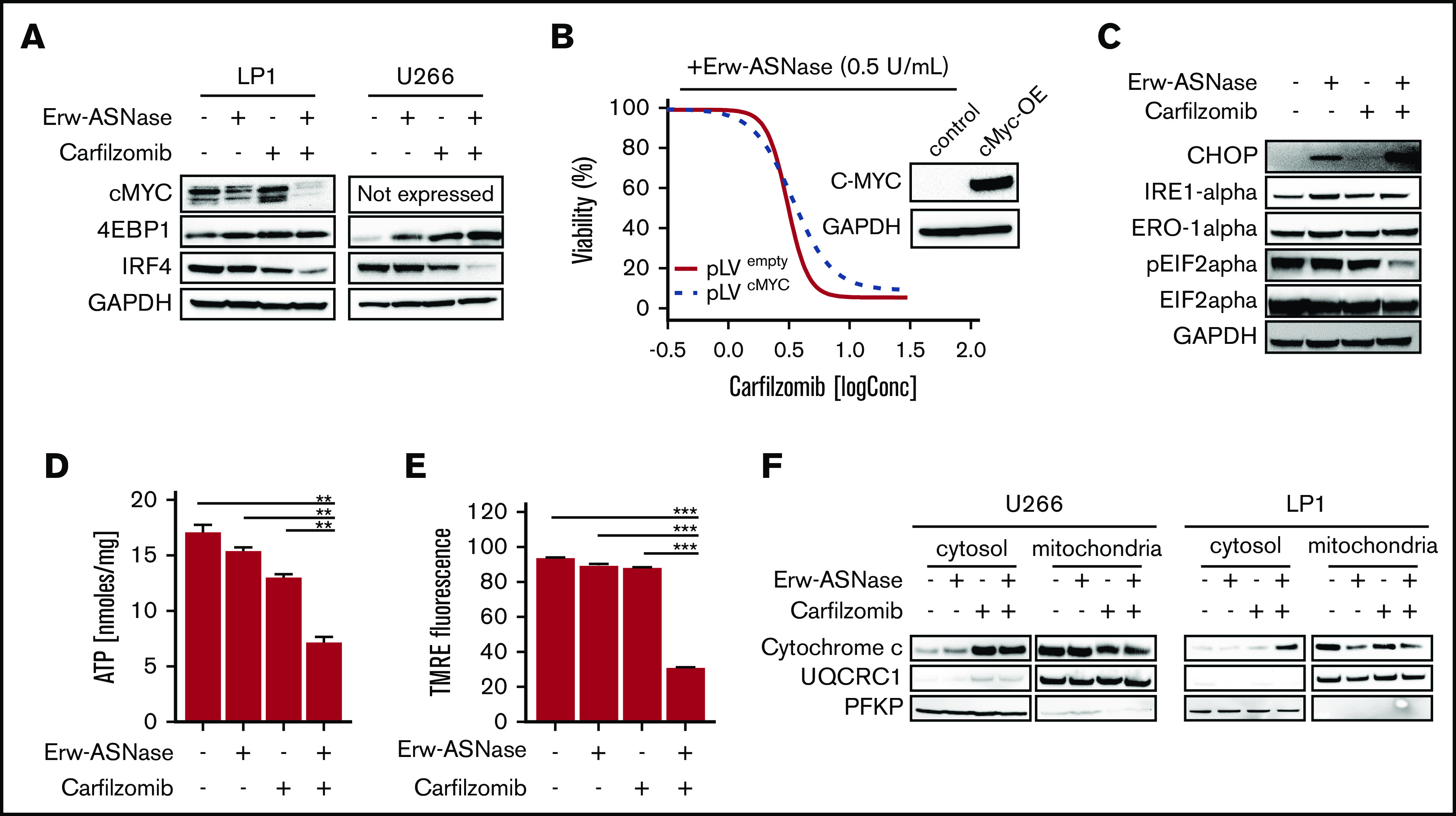
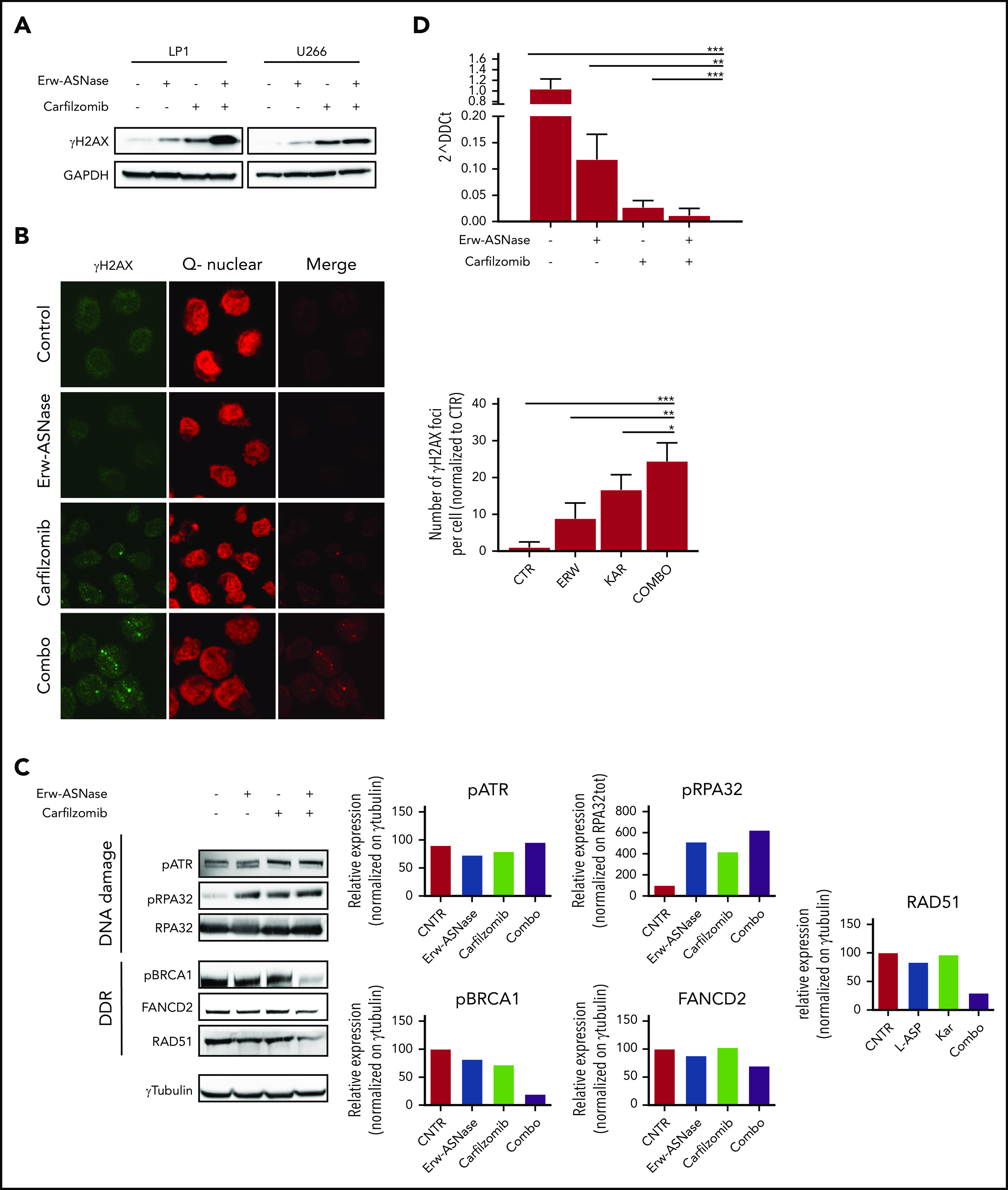
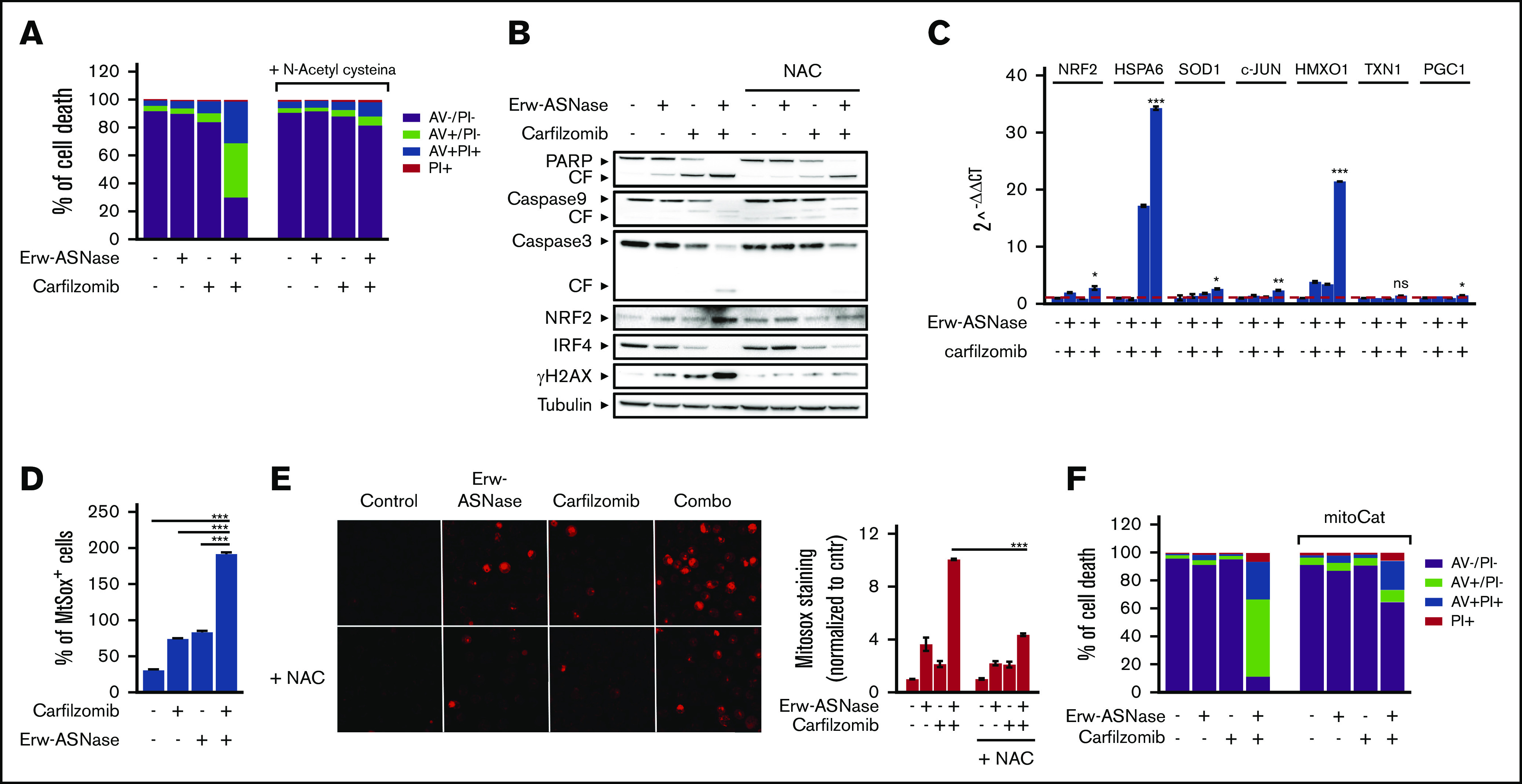
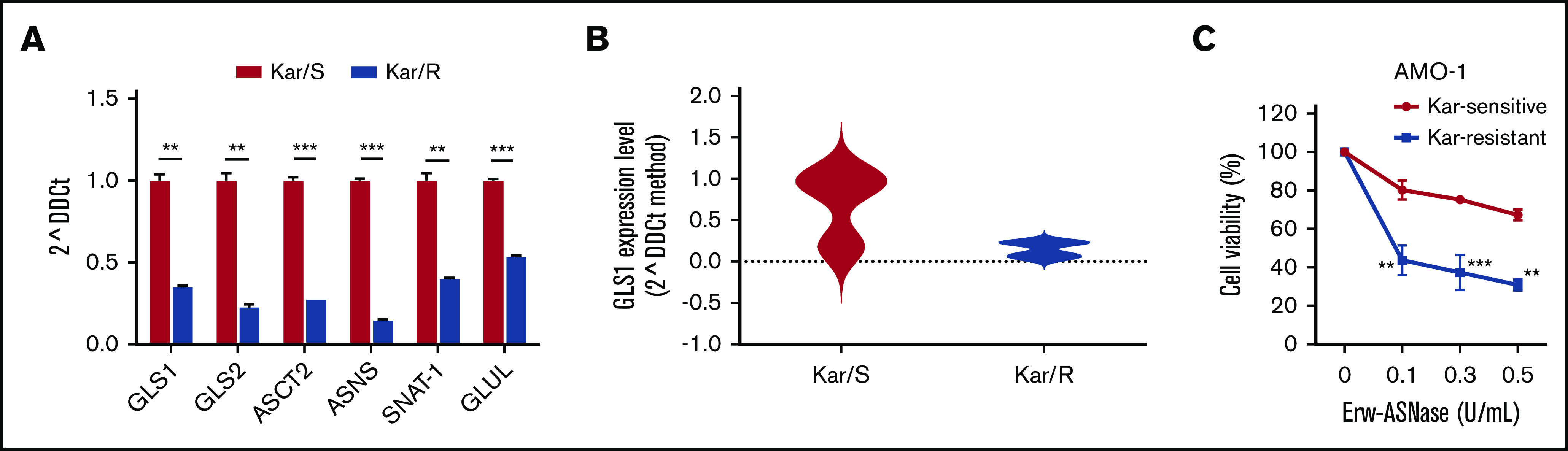
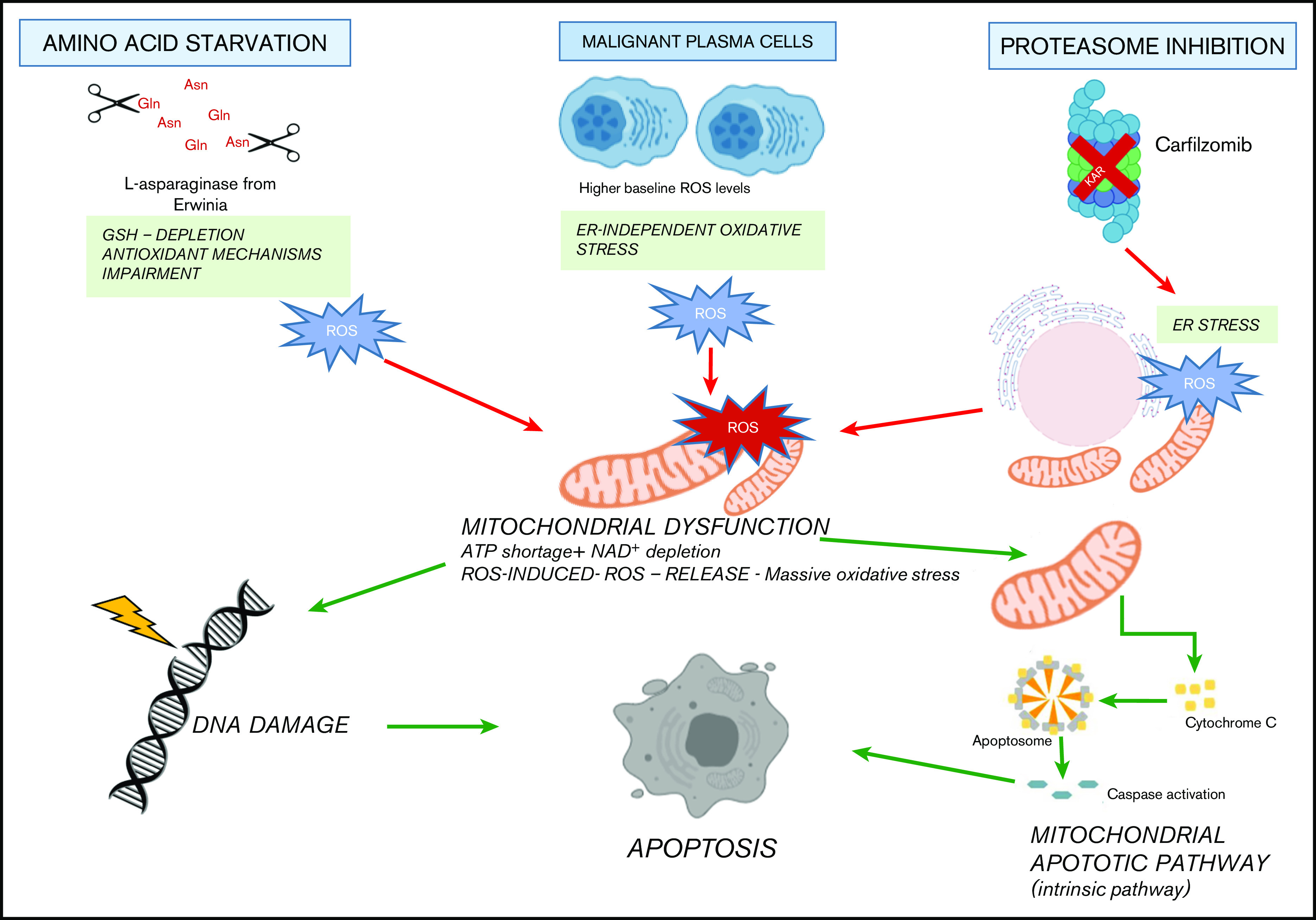
Metabolic reprogramming is emerging as a cancer vulnerability that could be therapeutically exploitable using different approaches, including amino acid depletion for those tumors that rely on exogenous amino acids for their maintenance. ʟ-Asparaginase (ASNase) has contributed to a significant improvement in acute lymphoblastic leukemia outcomes; however, toxicity and resistance limit its clinical use in other tumors. Here, we report that, in multiple myeloma (MM) cells, the DNA methylation status is significantly associated with reduced expression of ASNase-related gene signatures, thus suggesting ASNase sensitivity for this tumor. Therefore, we tested the effects of ASNase purified from Erwinia chrysanthemi (Erw-ASNase), combined with the next-generation proteasome inhibitor (PI) carfilzomib. We observed an impressive synergistic effect on MM cells, whereas normal peripheral blood mononuclear cells were not affected. Importantly, this effect was associated with increased reactive oxygen species (ROS) generation, compounded mitochondrial damage, and Nrf2 upregulation, regardless of the c-Myc oncogenic-specific program. Furthermore, the cotreatment resulted in genomic instability and DNA repair mechanism impairment via increased mitochondrial oxidative stress, which further enhanced its antitumor activity. Interestingly, carfilzomib-resistant cells were found to be highly dependent on amino acid starvation, as reflected by their higher sensitivity to Erw-ASNase treatment compared with isogenic cells. Overall, by affecting several cellular programs, Erw-ASNase makes MM cells more vulnerable to carfilzomib, providing proof of concept for clinical use of this combination as a novel strategy to enhance PI sensitivity in MM patients.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
