

Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome
- PMID: 32916129
- PMCID: PMC8109273
- DOI: 10.1016/j.cell.2020.08.007
Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome
Erratum in
-
Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome.Cell. 2020 Nov 12;183(4):1137-1140. doi: 10.1016/j.cell.2020.10.040. Cell. 2020. PMID: 33186523 No abstract available.
Abstract
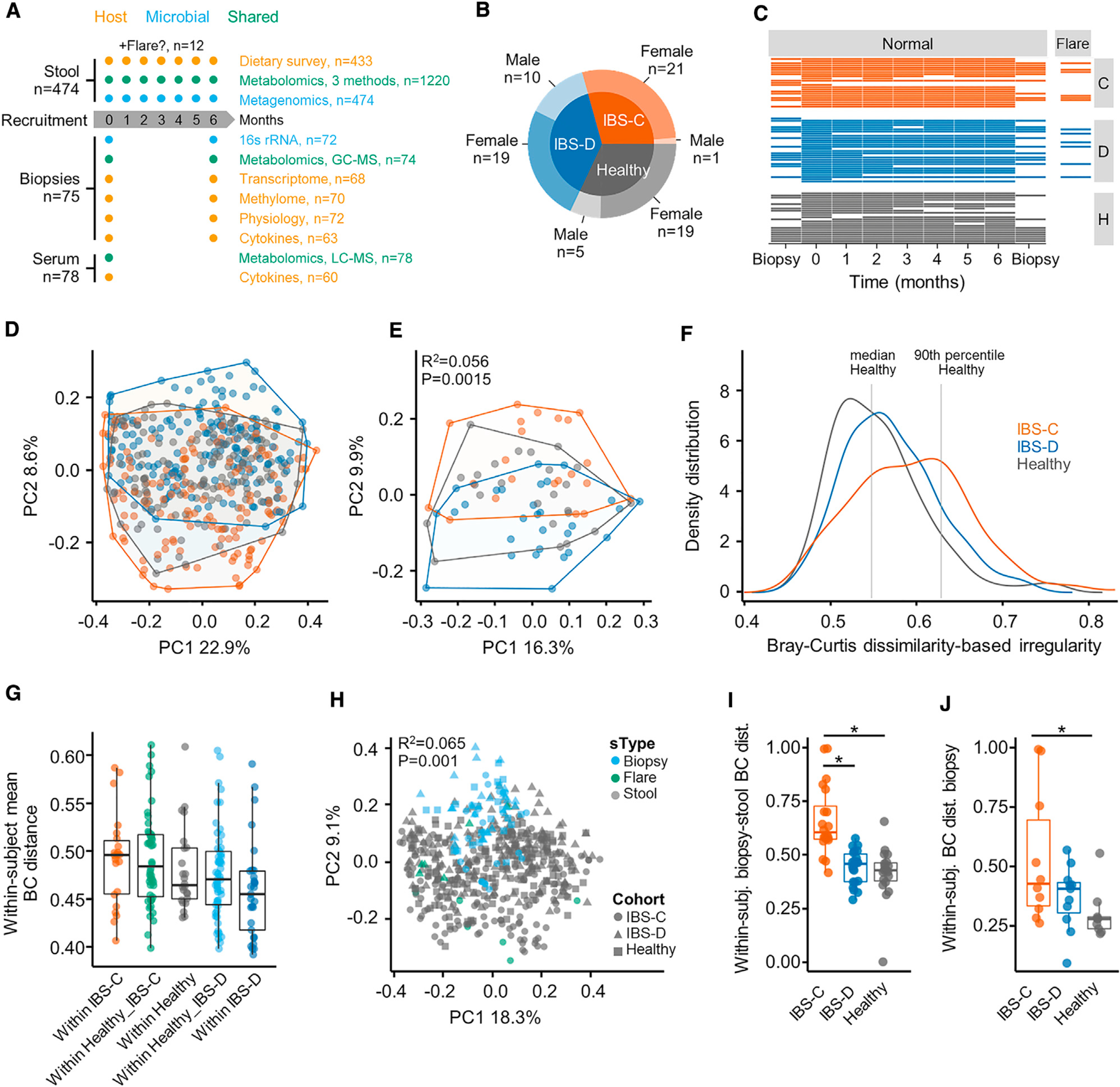
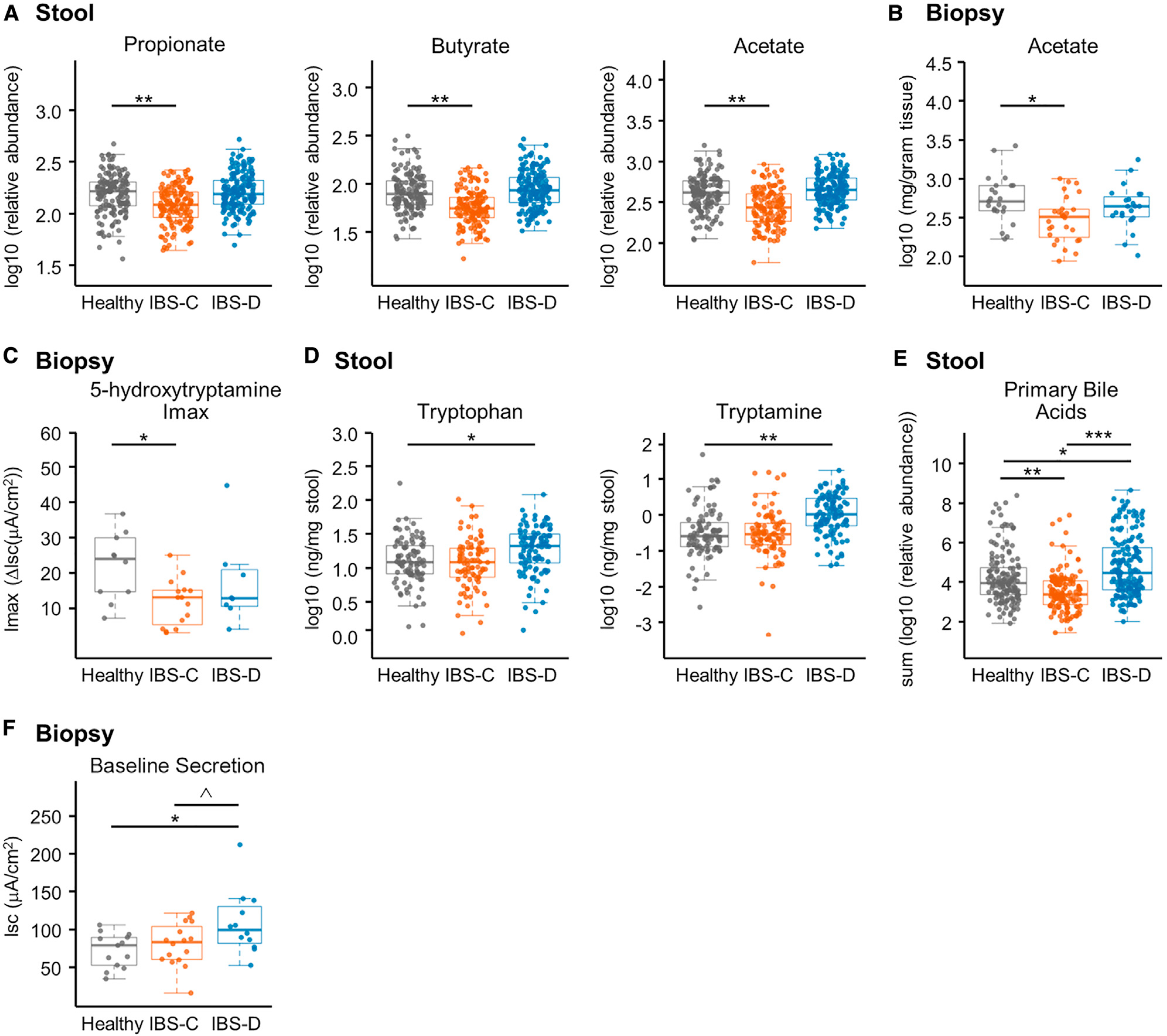
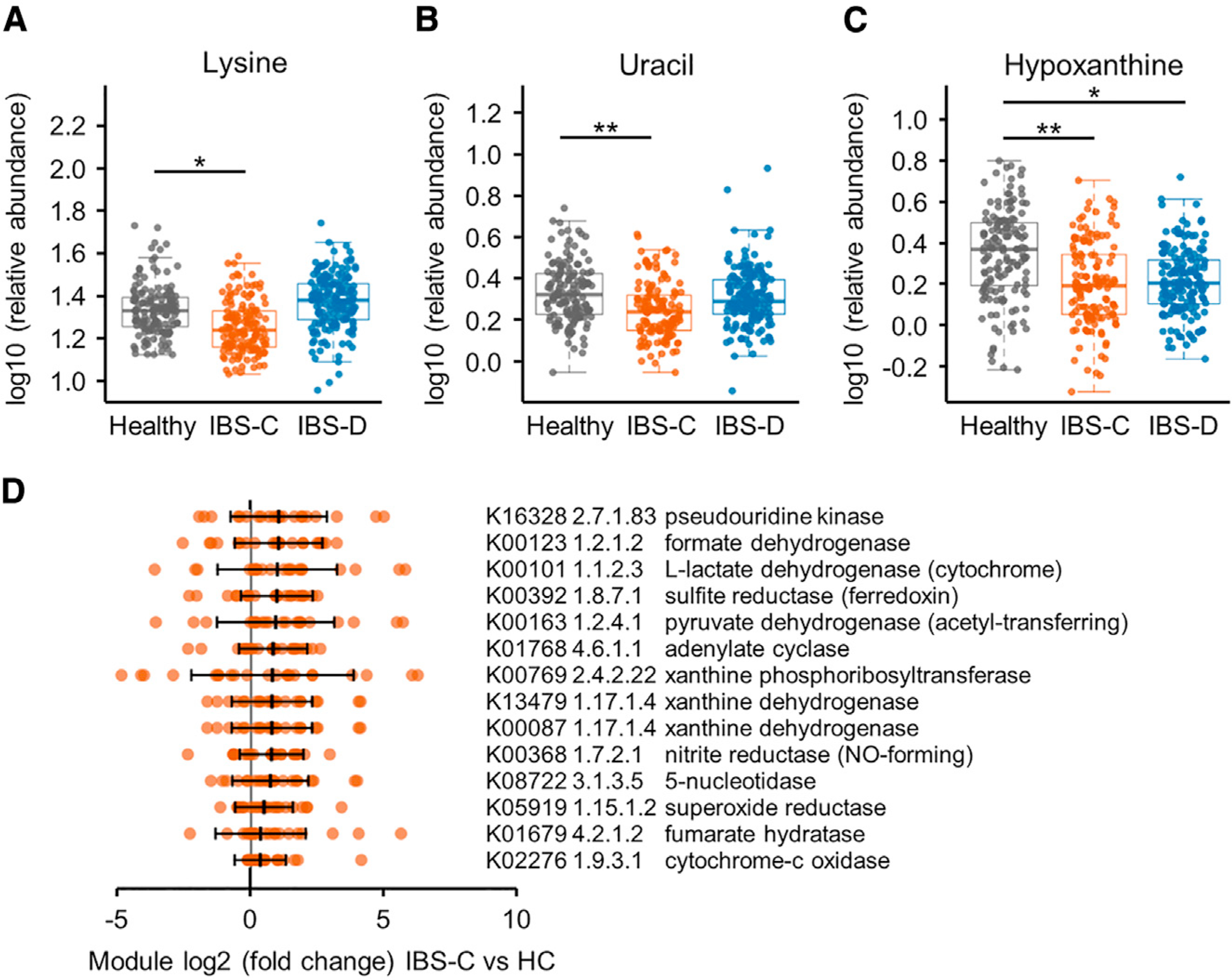
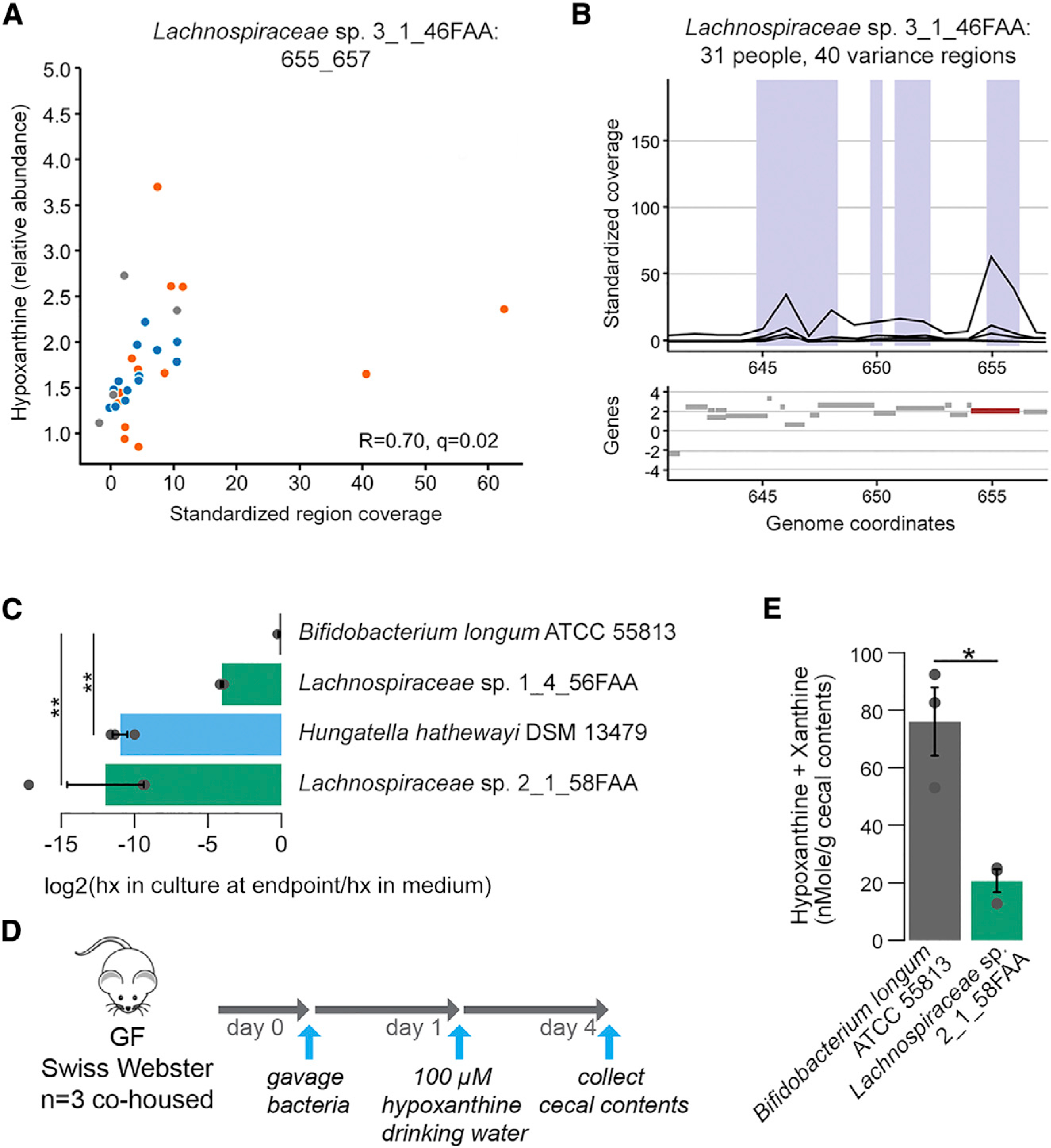
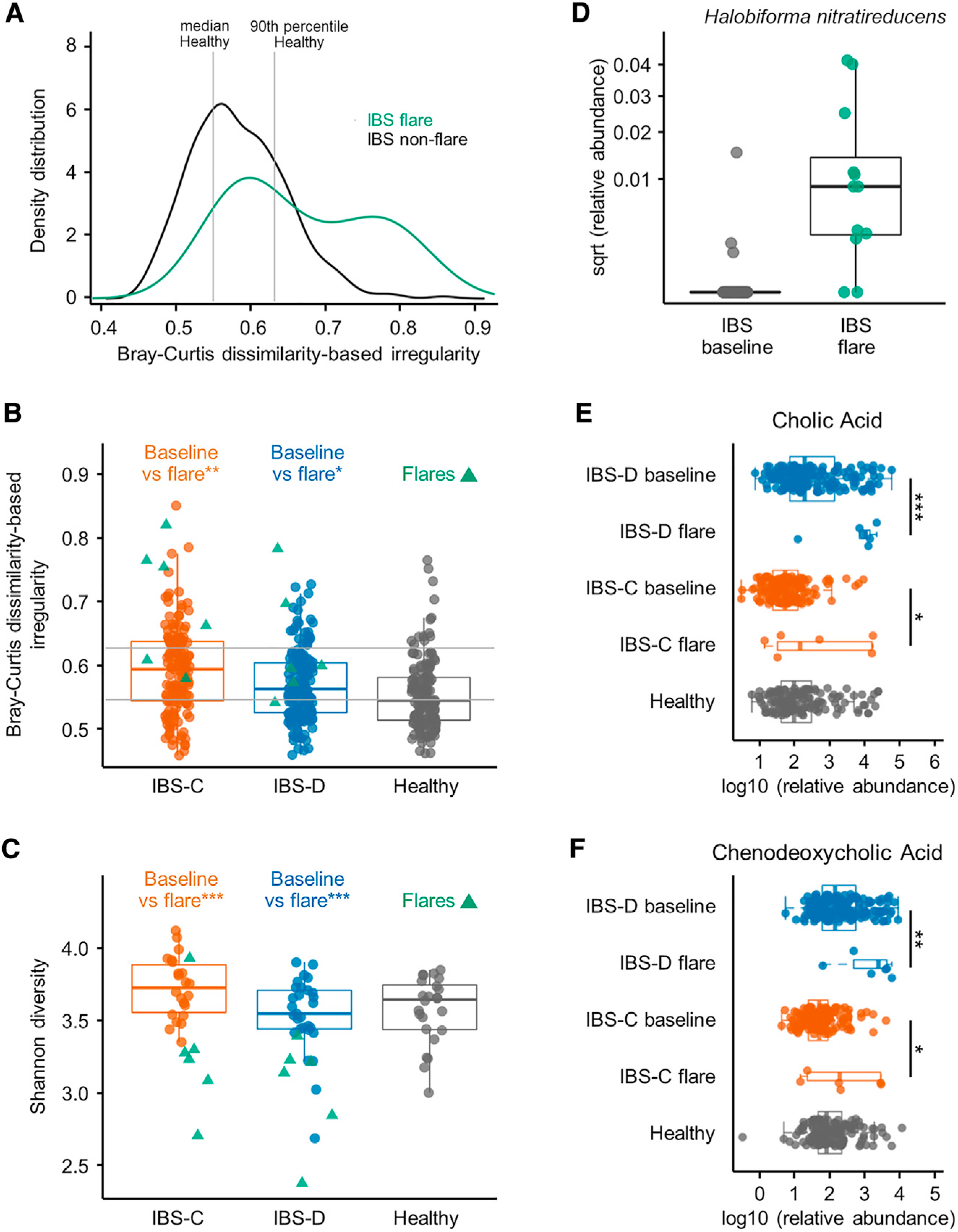
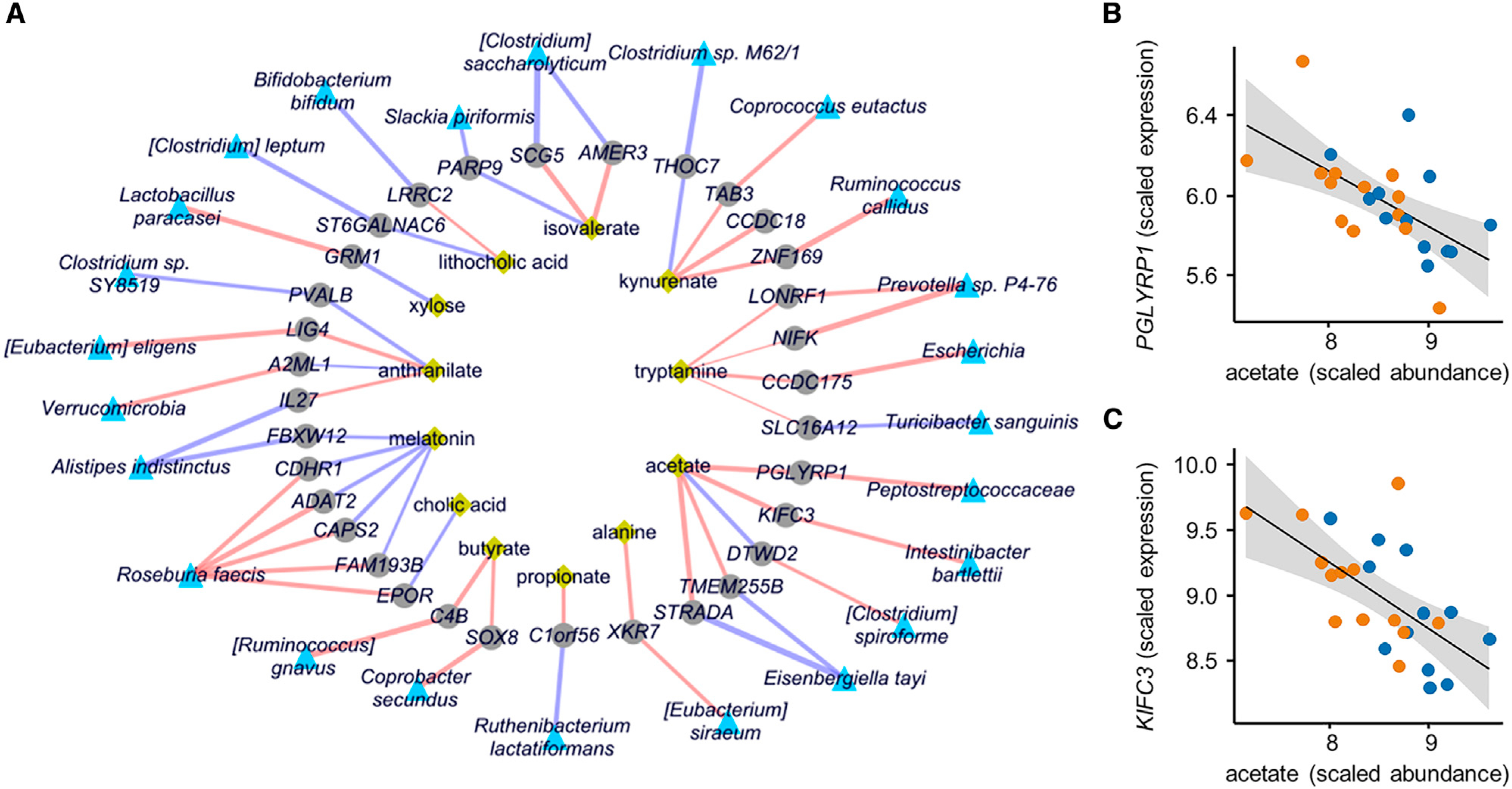
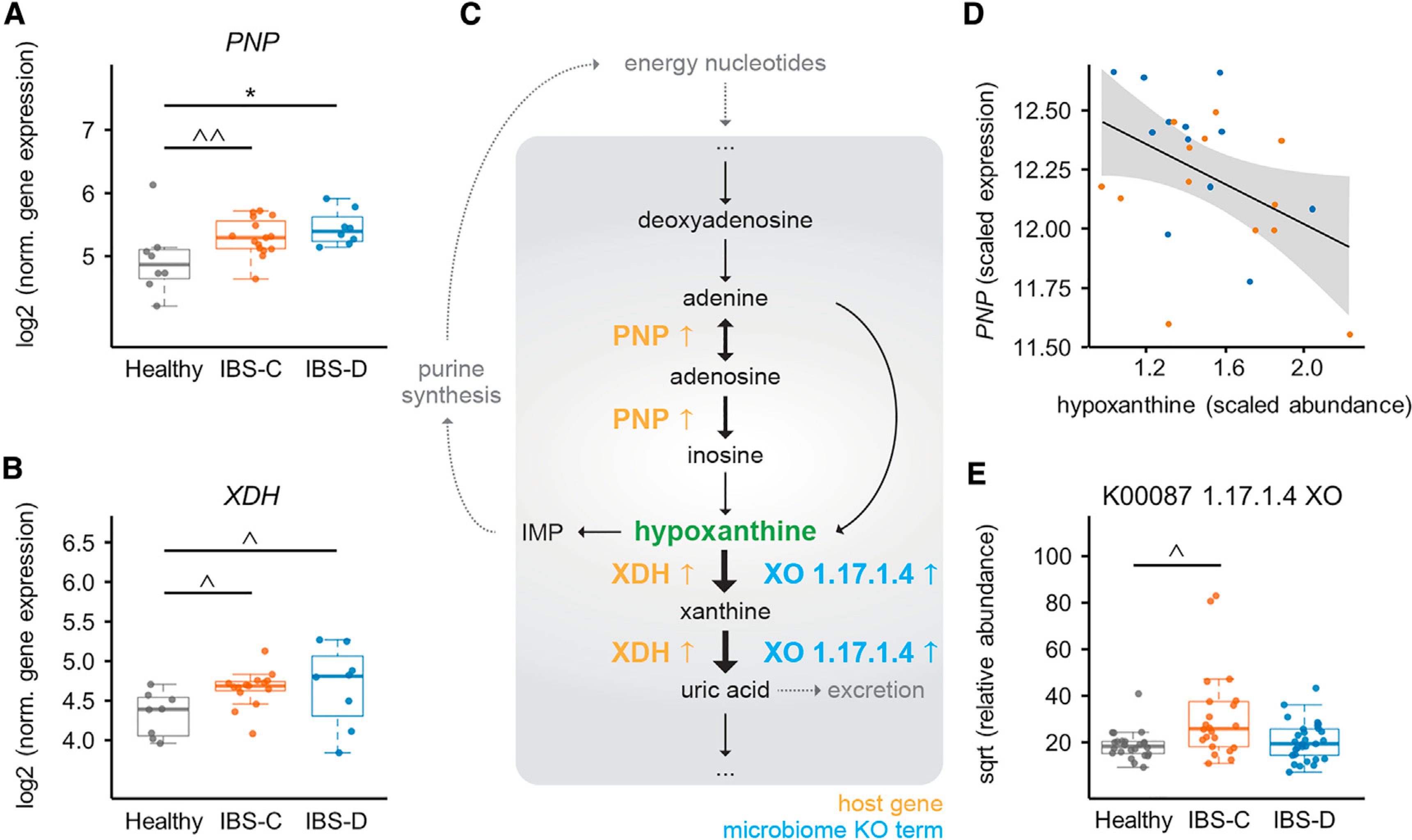
The gut microbiome has been implicated in multiple human chronic gastrointestinal (GI) disorders. Determining its mechanistic role in disease has been difficult due to apparent disconnects between animal and human studies and lack of an integrated multi-omics view of disease-specific physiological changes. We integrated longitudinal multi-omics data from the gut microbiome, metabolome, host epigenome, and transcriptome in the context of irritable bowel syndrome (IBS) host physiology. We identified IBS subtype-specific and symptom-related variation in microbial composition and function. A subset of identified changes in microbial metabolites correspond to host physiological mechanisms that are relevant to IBS. By integrating multiple data layers, we identified purine metabolism as a novel host-microbial metabolic pathway in IBS with translational potential. Our study highlights the importance of longitudinal sampling and integrating complementary multi-omics data to identify functional mechanisms that can serve as therapeutic targets in a comprehensive treatment strategy for chronic GI diseases. VIDEO ABSTRACT.
Keywords: bile acids; diet; functional bowel disorders; nucleosides; physiology; secretion; short chain fatti acids; symptom severity.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests P.C.K. is on the Advisory Board of Novome Biotechnologies and is an ad hoc consultant for Pendulum Therapeutics, IP group, and Otsuka Pharmaceuticals. P.C.K. holds patent US20170042860A1 for use of tryptamine producing bacteria (“Methods and materials for using Ruminococcus gnavus or Clostridium sporogenes to treat gastrointestinal disorders”), and P.C.K. and Mayo Clinic have a financial interest related to this research. These interests have been reviewed and managed in accordance with Mayo Clinic Conflict-of-Interest policies. D.B.K. serves as CEO of CoreBiome, a company involved in the commercialization of microbiome analysis and a wholly owned subsidiary of OraSure Technologies. These interests have been reviewed and managed by the University of Minnesota in accordance with its Conflict-of-Interest policies.
Figures
References
-
- Becker MA, Schumacher HR, MacDonald PA, Lloyd E, and Lademacher C (2009). Clinical efficacy and safety of successful longterm urate lowering with febuxostat or allopurinol in subjects with gout. J. Rheumatol 36, 1273–1282. - PubMed
-
- Bhattarai Y, Schmidt BA, Linden DR, Larson ED, Grover M, Beyder A, Farrugia G, and Kashyap PC (2017b). Human-derived gut microbiota modulates colonic secretion in mice by regulating 5-HT3 receptor expression via acetate production. Am. J. Physiol. Gastrointest. Liver Physiol 313, G80–G87. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
