

The role of autophagy in targeted therapy for acute myeloid leukemia
- PMID: 32917124
- PMCID: PMC8525957
- DOI: 10.1080/15548627.2020.1822628
The role of autophagy in targeted therapy for acute myeloid leukemia
Abstract
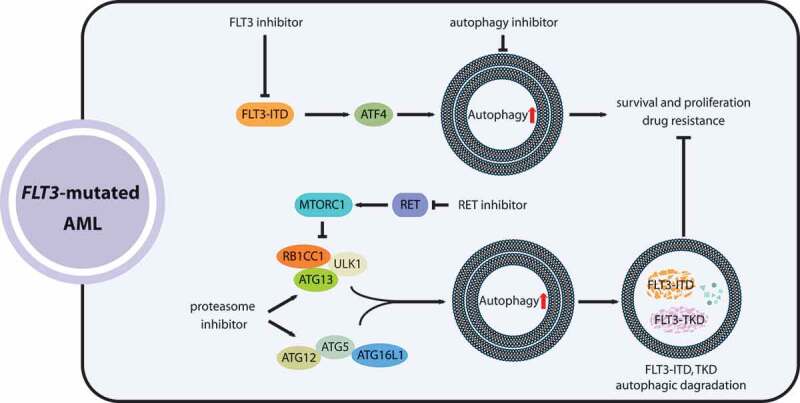
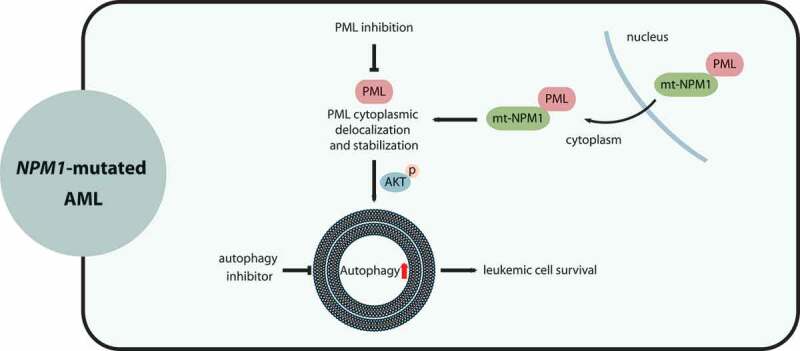
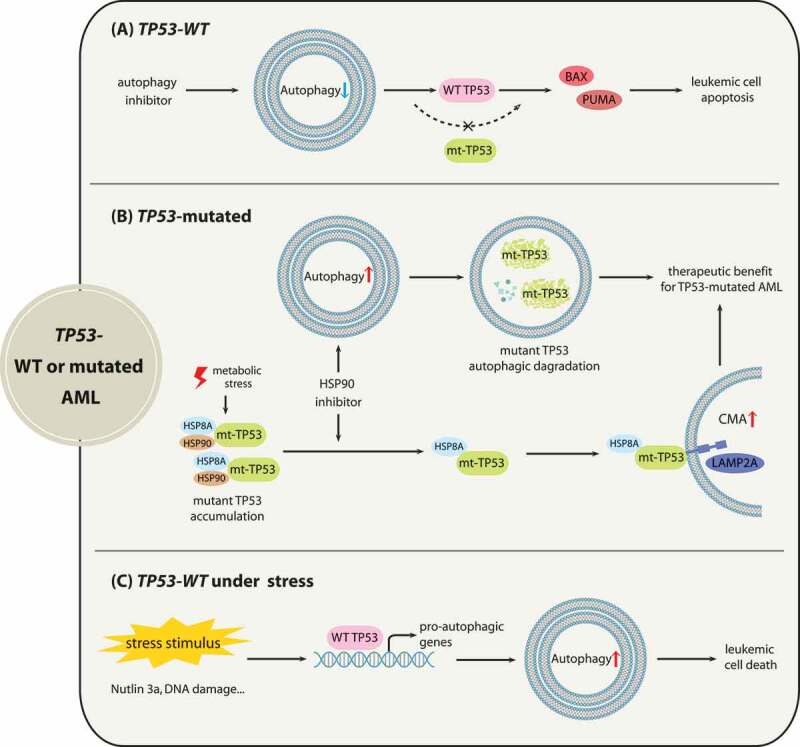
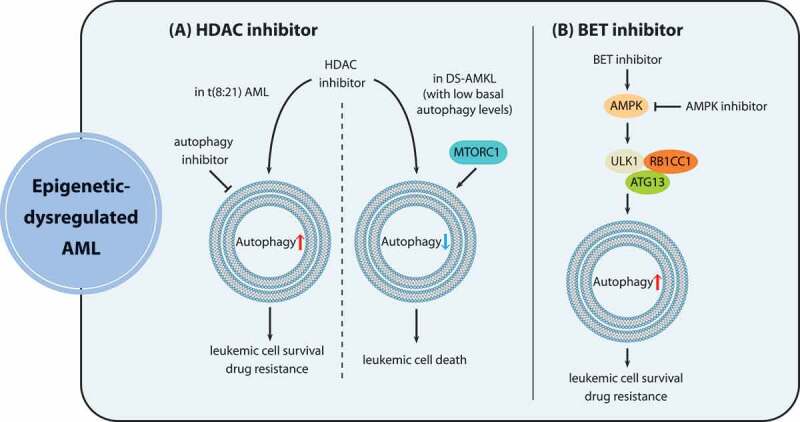
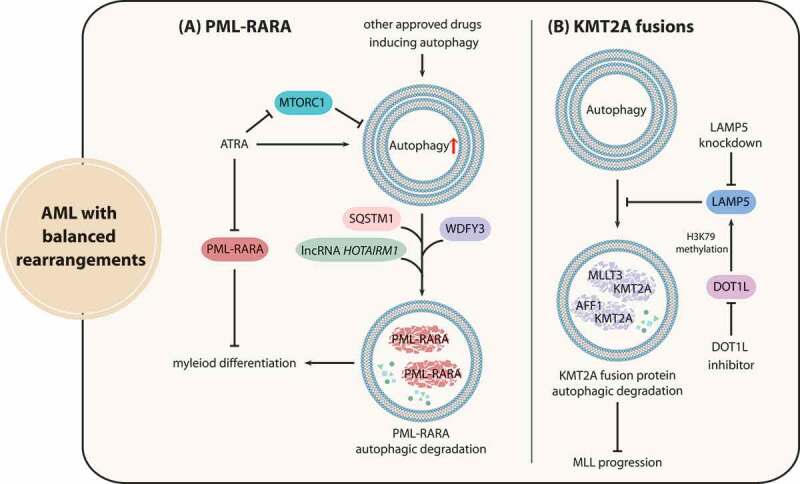
Although molecular targeted therapies have recently displayed therapeutic effects in acute myeloid leukemia (AML), limited response and acquired resistance remain common problems. Numerous studies have associated autophagy, an essential degradation process involved in the cellular response to stress, with the development and therapeutic response of cancers including AML. Thus, we review studies on the role of autophagy in AML development and summarize the linkage between autophagy and several recurrent genetic abnormalities in AML, highlighting the potential of capitalizing on autophagy modulation in targeted therapy for AML.Abbreviations: AML: acute myeloid leukemia; AMPK: AMP-activated protein kinase; APL: acute promyelocytic leukemia; ATG: autophagy related; ATM: ATM serine/threonine kinase; ATO: arsenic trioxide; ATRA: all trans retinoic acid; BCL2: BCL2 apoptosis regulator; BECN1: beclin 1; BET proteins, bromodomain and extra-terminal domain family; CMA: chaperone-mediated autophagy; CQ: chloroquine; DNMT, DNA methyltransferase; DOT1L: DOT1 like histone lysine methyltransferase; FLT3: fms related receptor tyrosine kinase 3; FIS1: fission, mitochondrial 1; HCQ: hydroxychloroquine; HSC: hematopoietic stem cell; IDH: isocitrate dehydrogenase; ITD: internal tandem duplication; KMT2A/MLL: lysine methyltransferase 2A; LSC: leukemia stem cell; MDS: myelodysplastic syndromes; MTORC1: mechanistic target of rapamycin kinase complex 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; NPM1: nucleophosmin 1; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PML: PML nuclear body scaffold; ROS: reactive oxygen species; RB1CC1/FIP200: RB1 inducible coiled-coil 1; SAHA: vorinostat; SQSTM1: sequestosome 1; TET2: tet methylcytosine dioxygenase 2; TKD: tyrosine kinase domain; TKI: tyrosine kinase inhibitor; TP53/p53: tumor protein p53; ULK1: unc-51 like autophagy activating kinase 1; VPA: valproic acid; WDFY3/ALFY: WD repeat and FYVE domain containing 3.
Keywords: Acute myeloid leukemia; autophagy; autophagy modulator; genetic abnormalities; molecular targeted therapy.
Conflict of interest statement
No potential conflict of interest was reported by the authors.
Figures
References
-
- Short NJ, Konopleva M, Kadia TM, et al. Advances in the treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov. 2020. Apr;10(4):506–525. - PubMed
-
- Antar AI, Otrock ZK, Jabbour E, et al. FLT3 inhibitors in acute myeloid leukemia: ten frequently asked questions. Leukemia. 2020. Jan 9;34(3):682–696. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous