

Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype
- PMID: 32917636
- PMCID: PMC7015694
- DOI: 10.1126/sciadv.aay9392
Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype
Abstract
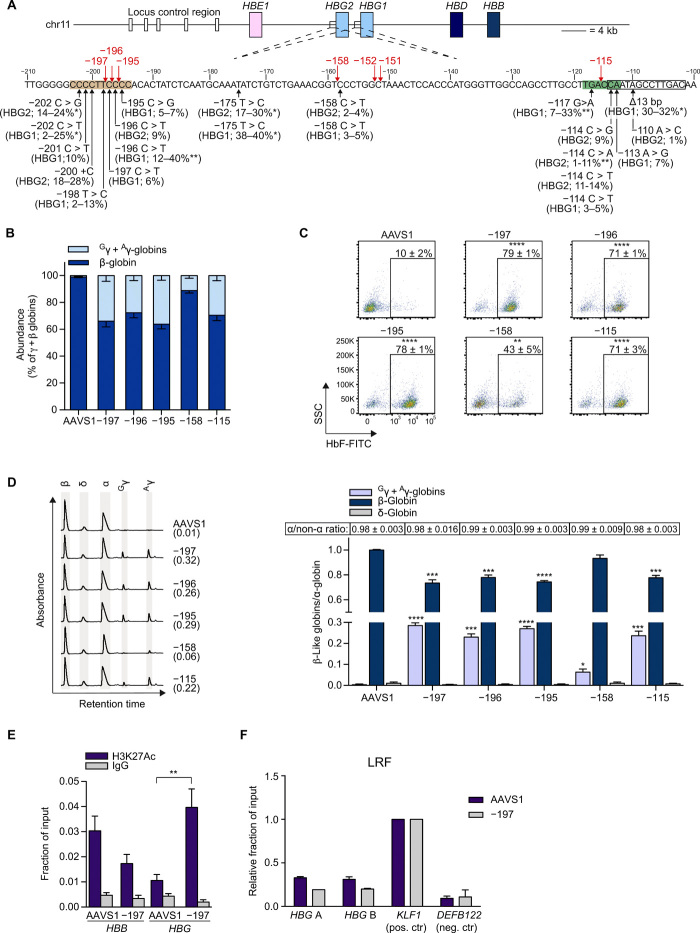
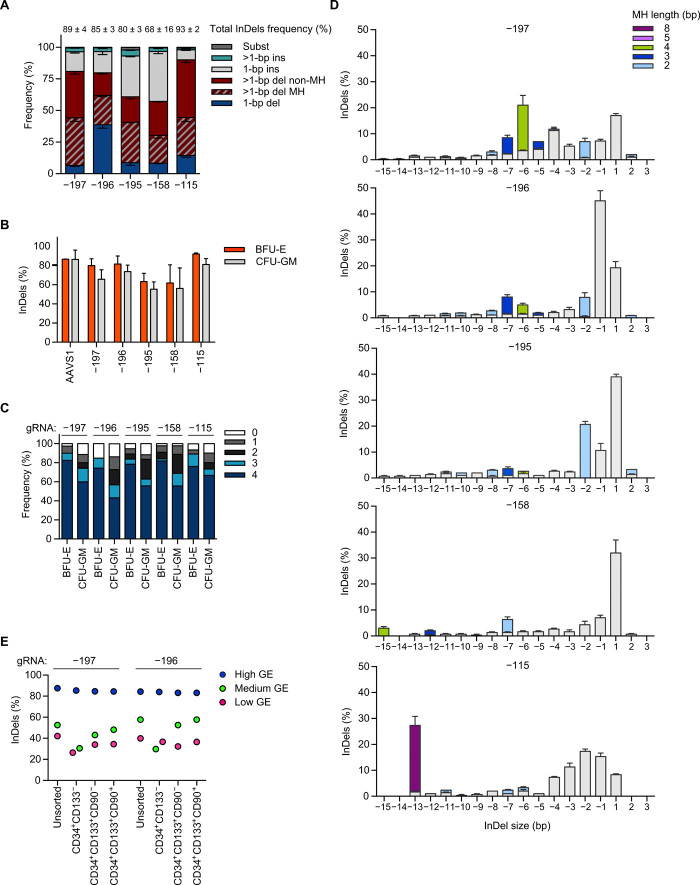
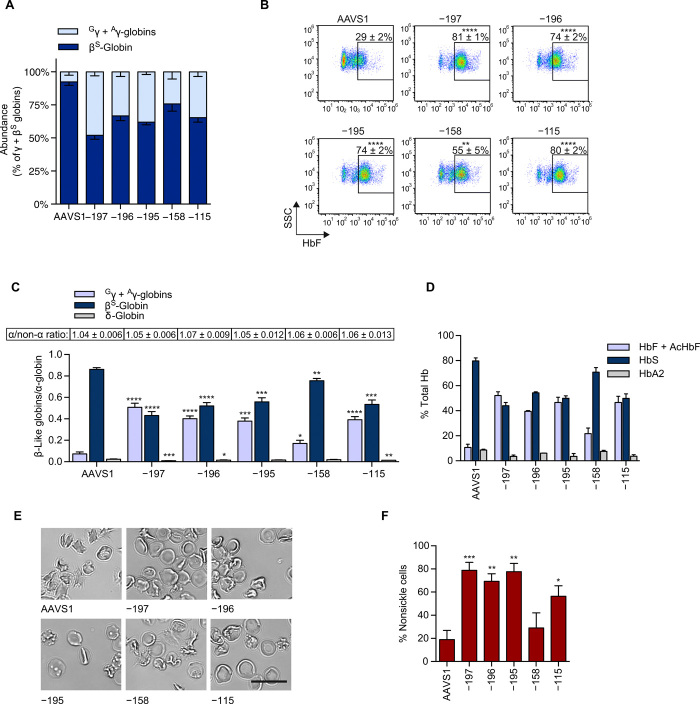
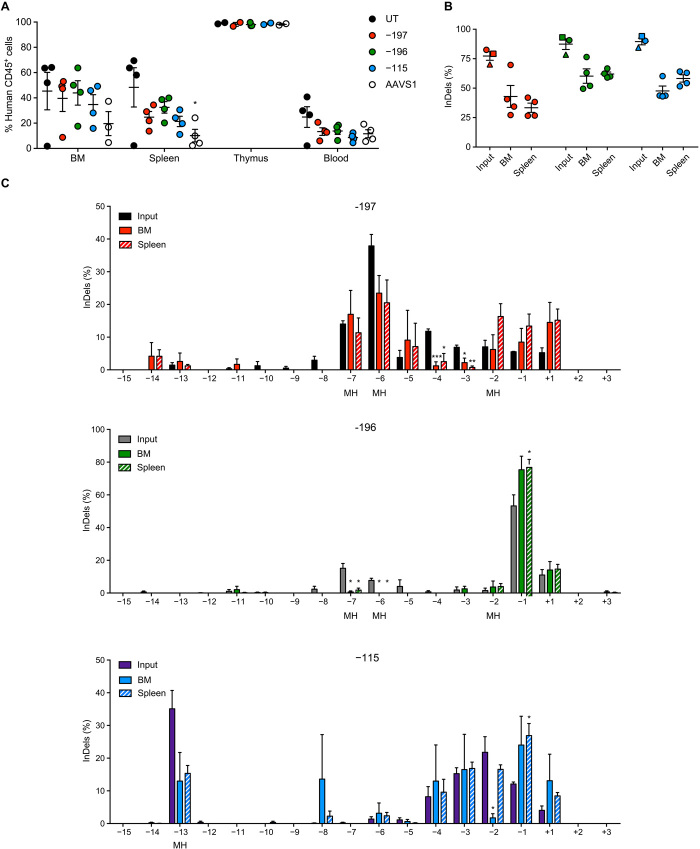
Sickle cell disease (SCD) is caused by a single amino acid change in the adult hemoglobin (Hb) β chain that causes Hb polymerization and red blood cell (RBC) sickling. The co-inheritance of mutations causing fetal γ-globin production in adult life hereditary persistence of fetal Hb (HPFH) reduces the clinical severity of SCD. HPFH mutations in the HBG γ-globin promoters disrupt binding sites for the repressors BCL11A and LRF. We used CRISPR-Cas9 to mimic HPFH mutations in the HBG promoters by generating insertions and deletions, leading to disruption of known and putative repressor binding sites. Editing of the LRF-binding site in patient-derived hematopoietic stem/progenitor cells (HSPCs) resulted in γ-globin derepression and correction of the sickling phenotype. Xenotransplantation of HSPCs treated with gRNAs targeting the LRF-binding site showed a high editing efficiency in repopulating HSPCs. This study identifies the LRF-binding site as a potent target for genome-editing treatment of SCD.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
Similar articles
-
Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal hemoglobin.Elife. 2022 Feb 11;11:e65421. doi: 10.7554/eLife.65421. Elife. 2022. PMID: 35147495 Free PMC article.
-
A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition.Nat Med. 2016 Sep;22(9):987-90. doi: 10.1038/nm.4170. Epub 2016 Aug 15. Nat Med. 2016. PMID: 27525524 Free PMC article.
-
Disrupting ZBTB7A or BCL11A binding sites reactivates fetal hemoglobin in erythroblasts from healthy and β0-thalassemia/HbE individuals.Sci Rep. 2025 Jul 15;15(1):25580. doi: 10.1038/s41598-025-10791-8. Sci Rep. 2025. PMID: 40665149 Free PMC article.
-
Transcriptional Repressor BCL11A in Erythroid Cells.Adv Exp Med Biol. 2024;1459:199-215. doi: 10.1007/978-3-031-62731-6_9. Adv Exp Med Biol. 2024. PMID: 39017845 Review.
-
Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies.Int J Mol Sci. 2023 May 31;24(11):9527. doi: 10.3390/ijms24119527. Int J Mol Sci. 2023. PMID: 37298481 Free PMC article. Review.
Cited by
-
Fetal hemoglobin in sickle cell anemia.Blood. 2020 Nov 19;136(21):2392-2400. doi: 10.1182/blood.2020007645. Blood. 2020. PMID: 32808012 Free PMC article. Review.
-
Induction of Fetal Hemoglobin by Introducing Natural Hereditary Persistence of Fetal Hemoglobin Mutations in the γ-Globin Gene Promoters for Genome Editing Therapies for β-Thalassemia.Front Genet. 2022 May 17;13:881937. doi: 10.3389/fgene.2022.881937. eCollection 2022. Front Genet. 2022. PMID: 35656314 Free PMC article.
-
Recent Approaches for Manipulating Globin Gene Expression in Treating Hemoglobinopathies.Front Genome Ed. 2021 Aug 2;3:618111. doi: 10.3389/fgeed.2021.618111. eCollection 2021. Front Genome Ed. 2021. PMID: 34713248 Free PMC article. Review.
-
CRISPR-Cas9 to induce fetal hemoglobin for the treatment of sickle cell disease.Mol Ther Methods Clin Dev. 2021 Oct 1;23:276-285. doi: 10.1016/j.omtm.2021.09.010. eCollection 2021 Dec 10. Mol Ther Methods Clin Dev. 2021. PMID: 34729375 Free PMC article. Review.
-
Multiplex base editing to protect from CD33 directed drugs for immune and gene therapy.Nat Commun. 2025 May 27;16(1):4899. doi: 10.1038/s41467-025-59713-2. Nat Commun. 2025. PMID: 40425554 Free PMC article.
References
-
- J. Kanter, J. F. Tisdale, J. L. Kwiatkowski, L. Krishnamurti, M. Y. Mapara, M. Schmidt, A. L. Miller, F. J. Pierciey Jr., W. Shi, J.-A. Ribeil, M. C. Walters, A. A. Thompson, Outcomes for initial patient cohorts with up to 33 months of follow-up in the Hgb-206 phase 1 trial, paper presented at the 60th ASH Annual Meeting, San Diego, CA, 1 to 4 December 2018.
-
- Thompson A. A., Walters M. C., Kwiatkowski J., Rasko J. E. J., Ribeil J. A., Hongeng S., Magrin E., Schiller G. J., Payen E., Semeraro M., Moshous D., Lefrere F., Puy H., Bourget P., Magnani A., Caccavelli L., Diana J. S., Suarez F., Monpoux F., Brousse V., Poirot C., Brouzes C., Meritet J. F., Pondarré C., Beuzard Y., Chrétien S., Lefebvre T., Teachey D. T., Anurathapan U., Ho P. J., von Kalle C., Kletzel M., Vichinsky E., Soni S., Veres G., Negre O., Ross R. W., Davidson D., Petrusich A., Sandler L., Asmal M., Hermine O., de Montalembert M., Hacein-Bey-Abina S., Blanche S., Leboulch P., Cavazzana M., Gene therapy in patients with transfusion-dependent β-Thalassemia. N. Engl. J. Med. 378, 1479–1493 (2018). - PubMed
-
- Marktel S., Scaramuzza S., Cicalese M. P., Giglio F., Galimberti S., Lidonnici M. R., Calbi V., Assanelli A., Bernardo M. E., Rossi C., Calabria A., Milani R., Gattillo S., Benedicenti F., Spinozzi G., Aprile A., Bergami A., Casiraghi M., Consiglieri G., Masera N., D’Angelo E., Mirra N., Origa R., Tartaglione I., Perrotta S., Winter R., Coppola M., Viarengo G., Santoleri L., Graziadei G., Gabaldo M., Valsecchi M. G., Montini E., Naldini L., Cappellini M. D., Ciceri F., Aiuti A., Ferrari G., Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent β-thalassemia. Nat. Med. 25, 234–241 (2019). - PubMed
-
- Ribeil J. A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., el Nemer W., Bartolucci P., Weber L., Puy H., Meritet J. F., Grevent D., Beuzard Y., Chrétien S., Lefebvre T., Ross R. W., Negre O., Veres G., Sandler L., Soni S., de Montalembert M., Blanche S., Leboulch P., Cavazzana M., Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 376, 848–855 (2017). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
