

Incomplete annotation has a disproportionate impact on our understanding of Mendelian and complex neurogenetic disorders
- PMID: 32917675
- PMCID: PMC7286675
- DOI: 10.1126/sciadv.aay8299
Incomplete annotation has a disproportionate impact on our understanding of Mendelian and complex neurogenetic disorders
Abstract
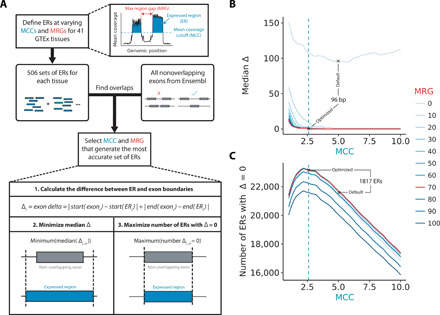
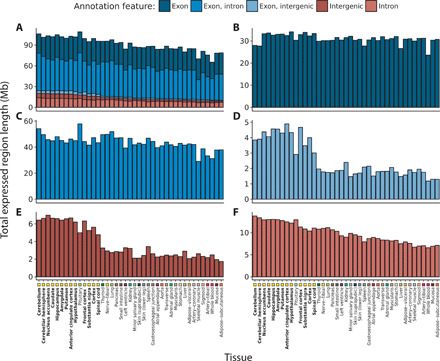
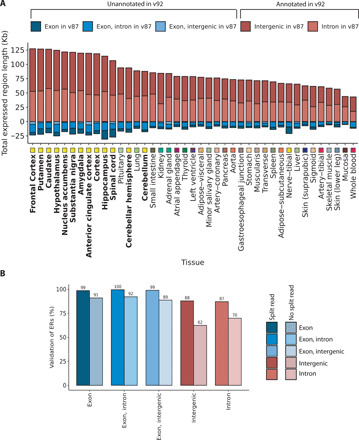
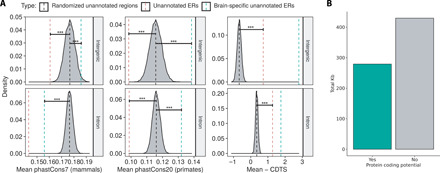
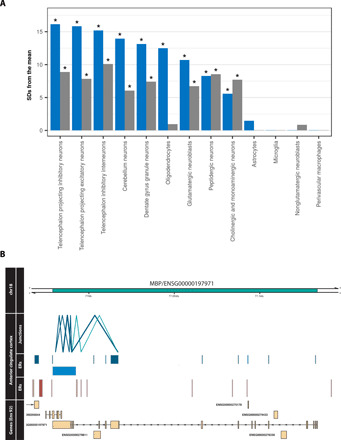
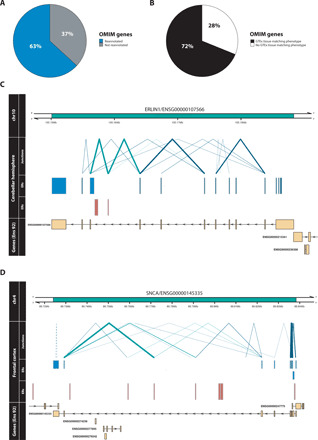
Growing evidence suggests that human gene annotation remains incomplete; however, it is unclear how this affects different tissues and our understanding of different disorders. Here, we detect previously unannotated transcription from Genotype-Tissue Expression RNA sequencing data across 41 human tissues. We connect this unannotated transcription to known genes, confirming that human gene annotation remains incomplete, even among well-studied genes including 63% of the Online Mendelian Inheritance in Man-morbid catalog and 317 neurodegeneration-associated genes. We find the greatest abundance of unannotated transcription in brain and genes highly expressed in brain are more likely to be reannotated. We explore examples of reannotated disease genes, such as SNCA, for which we experimentally validate a previously unidentified, brain-specific, potentially protein-coding exon. We release all tissue-specific transcriptomes through vizER: http://rytenlab.com/browser/app/vizER We anticipate that this resource will facilitate more accurate genetic analysis, with the greatest impact on our understanding of Mendelian and complex neurogenetic disorders.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution License 4.0 (CC BY).
Figures
References
-
- Harrow J., Frankish A., Gonzalez J. M., Tapanari E., Diekhans M., Kokocinski F., Aken B. L., Barrell D., Zadissa A., Searle S., Barnes I., Bignell A., Boychenko V., Hunt T., Kay M., Mukherjee G., Rajan J., Despacio-Reyes G., Saunders G., Steward C., Harte R., Lin M., Howald C., Tanzer A., Derrien T., Chrast J., Walters N., Balasubramanian S., Pei B., Tress M., Rodriguez J. M., Ezkurdia I., van Baren J., Brent M., Haussler D., Kellis M., Valencia A., Reymond A., Gerstein M., Guigo R., Hubbard T. J., GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012). - PMC - PubMed
-
- O’Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., Veigh R. M., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., Astashyn A., Badretdin A., Bao Y., Blinkova O., Brover V., Chetvernin V., Choi J., Cox E., Ermolaeva O., Farrell C. M., Goldfarb T., Gupta T., Haft D., Hatcher E., Hlavina W., Joardar V. S., Kodali V. K., Li W., Maglott D., Masterson P., Mc Garvey K. M., Murphy M. R., O’Neill K., Pujar S., Rangwala S. H., Rausch D., Riddick L. D., Schoch C., Shkeda A., Storz S. S., Sun H., Thibaud-Nissen F., Tolstoy I., Tully R. E., Vatsan A. R., Wallin C., Webb D., Wu W., Landrum M. J., Kimchi A., Tatusova T., Cuccio M. D., Kitts P., Murphy T. D., Pruitt K. D., Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016). - PMC - PubMed
-
- Zerbino D. R., Achuthan P., Akanni W., Amode M. R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C. G., Gil L., Gordon L., Haggerty L., Haskell E., Hourlier T., Izuogu O. G., Janacek S. H., Juettemann T., To J. K., Laird M. R., Lavidas I., Liu Z., Loveland J. E., Maurel T., McLaren W., Moore B., Mudge J., Murphy D. N., Newman V., Nuhn M., Ogeh D., Ong C. K., Parker A., Patricio M., Riat H. S., Schuilenburg H., Sheppard D., Sparrow H., Taylor K., Thormann A., Vullo A., Walts B., Zadissa A., Frankish A., Hunt S. E., Kostadima M., Langridge N., Martin F. J., Muffato M., Perry E., Ruffier M., Staines D. M., Trevanion S. J., Aken B. L., Cunningham F., Yates A., Flicek P., Ensembl 2018. Nucleic Acids Res. 46, D754–D761 (2018). - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
