

Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging
- PMID: 32917871
- PMCID: PMC7486940
- DOI: 10.1038/s41467-020-18405-9
Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging
Erratum in
-
Publisher Correction: Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging.Nat Commun. 2021 Apr 9;12(1):2271. doi: 10.1038/s41467-021-22789-7. Nat Commun. 2021. PMID: 33837216 Free PMC article. No abstract available.
Abstract
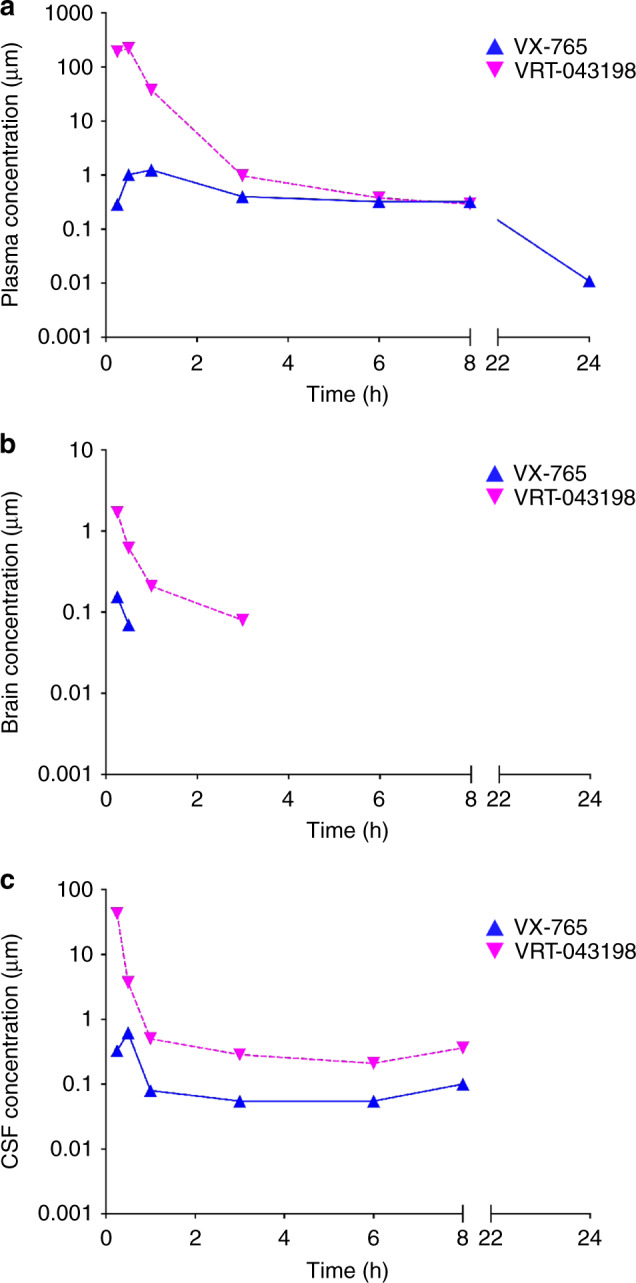
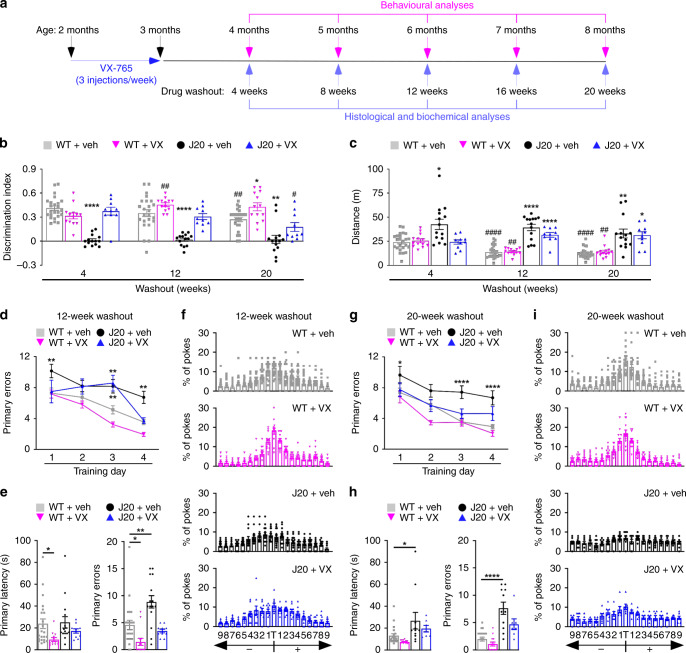
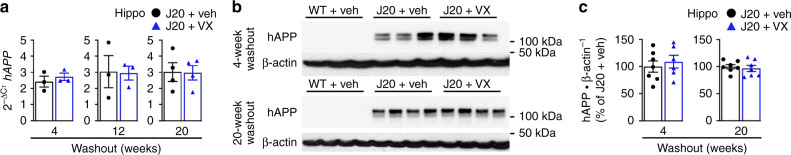
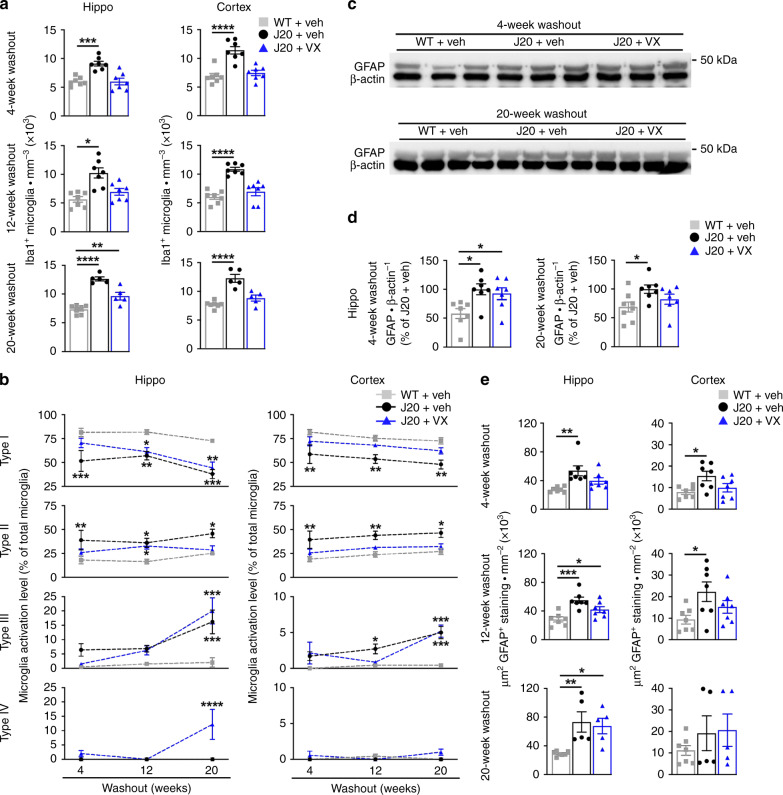
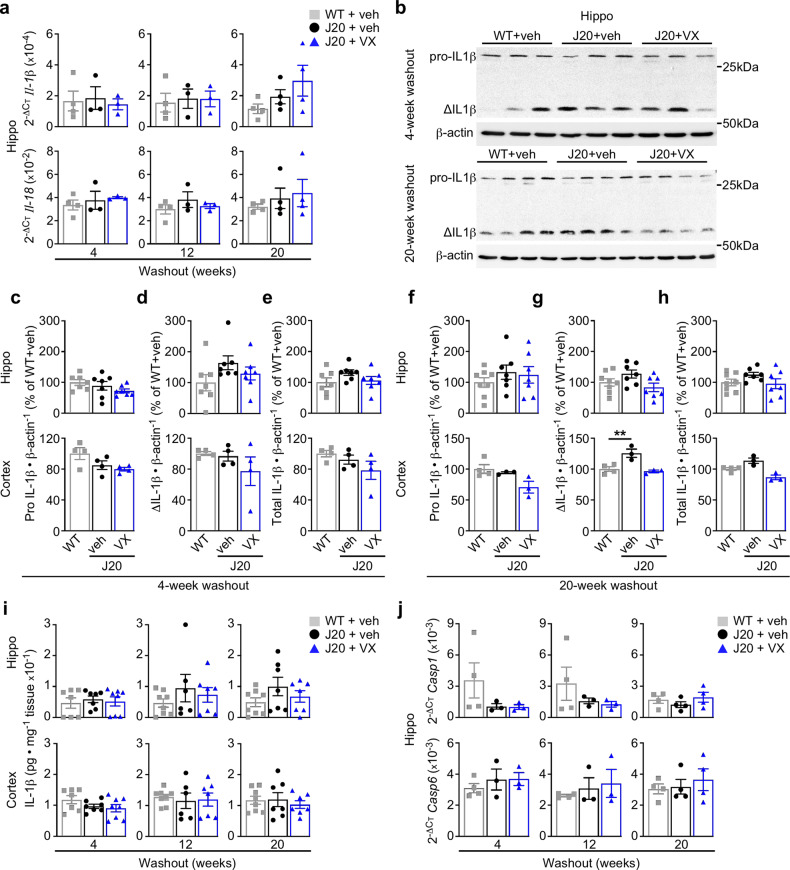
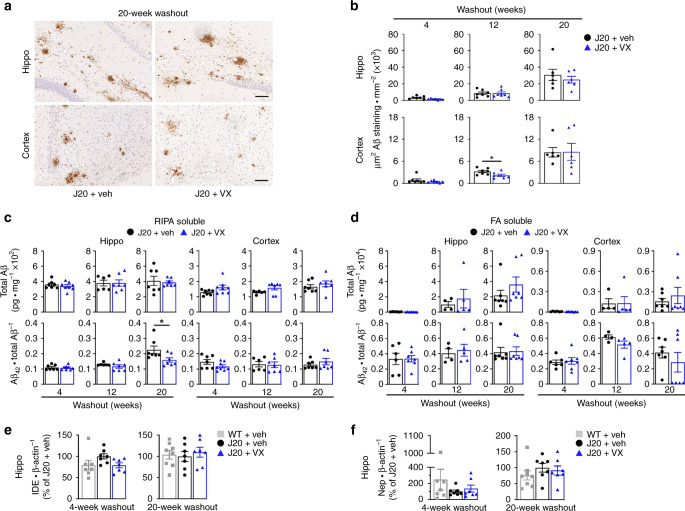
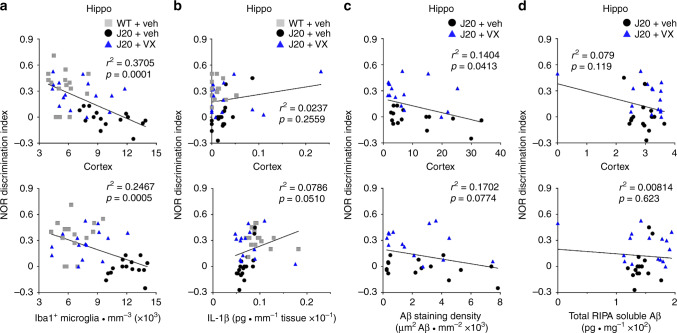
Early therapeutic interventions are essential to prevent Alzheimer Disease (AD). The association of several inflammation-related genetic markers with AD and the early activation of pro-inflammatory pathways in AD suggest inflammation as a plausible therapeutic target. Inflammatory Caspase-1 has a significant impact on AD-like pathophysiology and Caspase-1 inhibitor, VX-765, reverses cognitive deficits in AD mouse models. Here, a one-month pre-symptomatic treatment of Swedish/Indiana mutant amyloid precursor protein (APPSw/Ind) J20 and wild-type mice with VX-765 delays both APPSw/Ind- and age-induced episodic and spatial memory deficits. VX-765 delays inflammation without considerably affecting soluble and aggregated amyloid beta peptide (Aβ) levels. Episodic memory scores correlate negatively with microglial activation. These results suggest that Caspase-1-mediated inflammation occurs early in the disease and raise hope that VX-765, a previously Food and Drug Administration-approved drug for human CNS clinical trials, may be a useful drug to prevent the onset of cognitive deficits and brain inflammation in AD.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
