

Pathomechanisms and Clinical Implications of Myasthenic Syndromes Exacerbated and Induced by Medical Treatments
- PMID: 32922263
- PMCID: PMC7457047
- DOI: 10.3389/fnmol.2020.00156
Pathomechanisms and Clinical Implications of Myasthenic Syndromes Exacerbated and Induced by Medical Treatments
Abstract
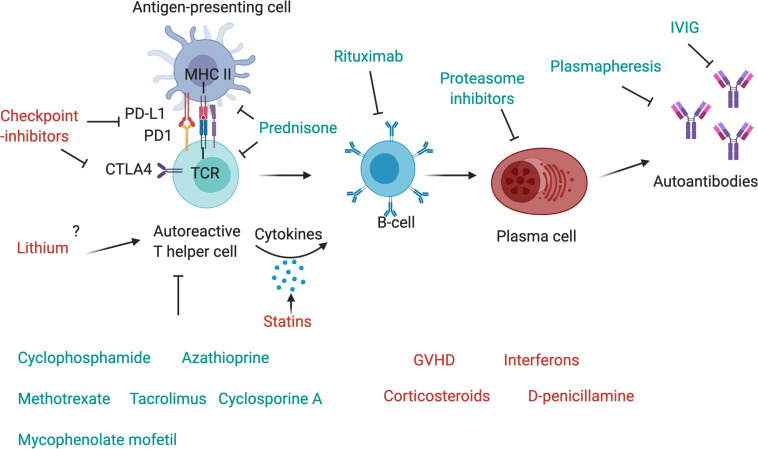
Myasthenic syndromes are typically characterized by muscle weakness and increased fatigability due to an impaired transmission at the neuromuscular junction (NMJ). Most cases are caused by acquired autoimmune conditions such as myasthenia gravis (MG), typically with antibodies against the acetylcholine receptor (AChR). Different drugs are among the major factors that may complicate pre-existing autoimmune myasthenic conditions by further impairing transmission at the NMJ. Some clinical observations are substantiated by experimental data, indicating that presynaptic, postsynaptic or more complex pathomechanisms at the NMJ may be involved, depending on the individual compound. Most robust data exist for the risks associated with some antibiotics (e.g., aminoglycosides, ketolides, fluoroquinolones) and cardiovascular medications (e.g., class Ia antiarrhythmics, beta blockers). Apart from primarily autoimmune-mediated disorders of the NMJ, de novo myasthenic manifestations may also be triggered by medical treatments that induce an autoimmune reaction. Most notably, there is growing evidence that the immune checkpoint inhibitors (ICI), a modern class of drugs to treat various malignancies, represent a relevant risk factor to develop severe and progressive medication-induced myasthenia via an immune-mediated mechanism. From a clinical perspective, it is of utmost importance for the treating physicians to be aware of such adverse treatment effects and their consequences. In this article, we aim to summarize existing evidence regarding the key molecular and immunological mechanisms as well as the clinical implications of medication-aggravated and medication-induced myasthenic syndromes.
Keywords: drug-induced myasthenia; drug-related myasthenia; immune checkpoint inhibitors; neuromuscular junction; neuromuscular transmission.
Copyright © 2020 Krenn, Grisold, Wohlfarth, Rath, Cetin, Koneczny and Zimprich.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources