

Potential protease inhibitors and their combinations to block SARS-CoV-2
- PMID: 32924827
- PMCID: PMC7544937
- DOI: 10.1080/07391102.2020.1819881
Potential protease inhibitors and their combinations to block SARS-CoV-2
Abstract
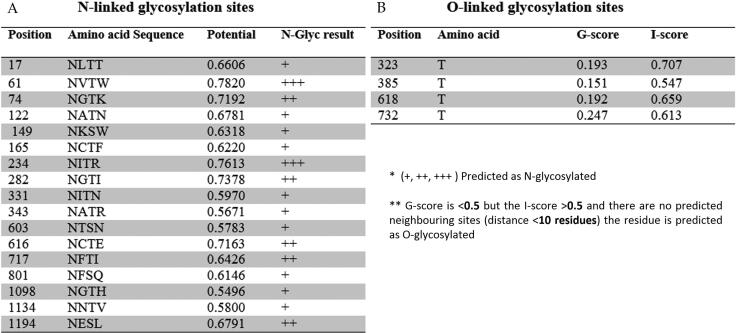
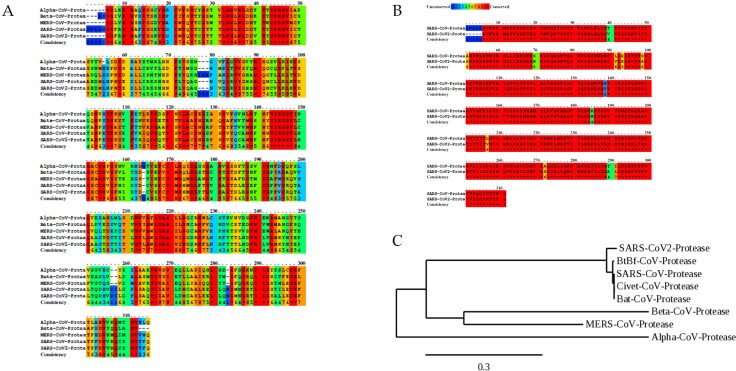
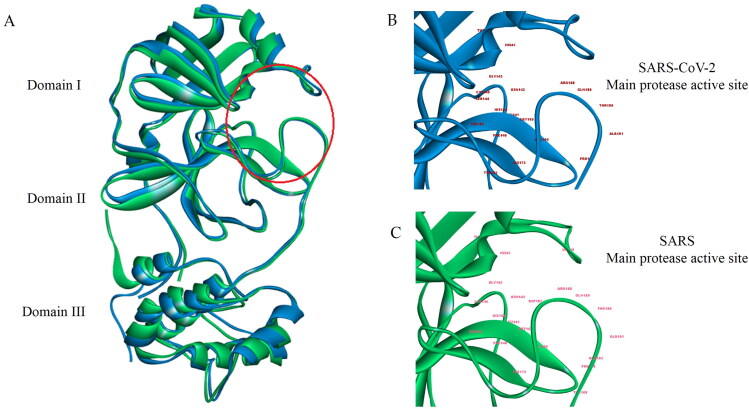
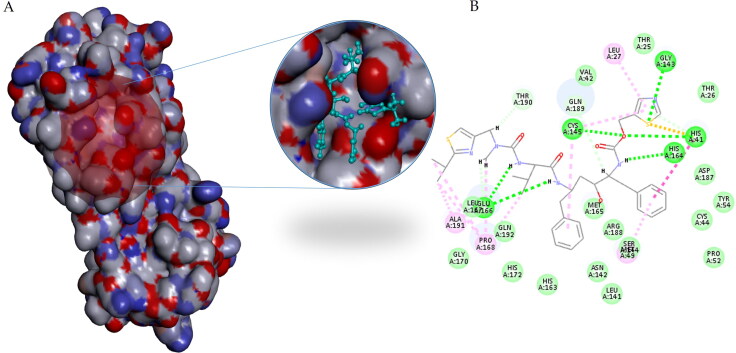
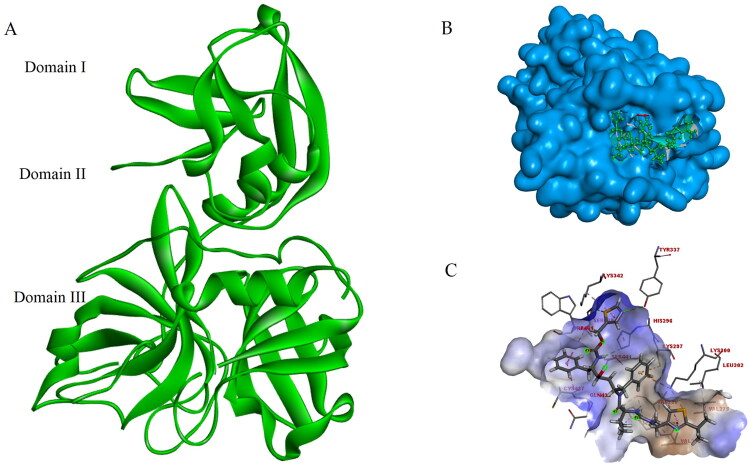
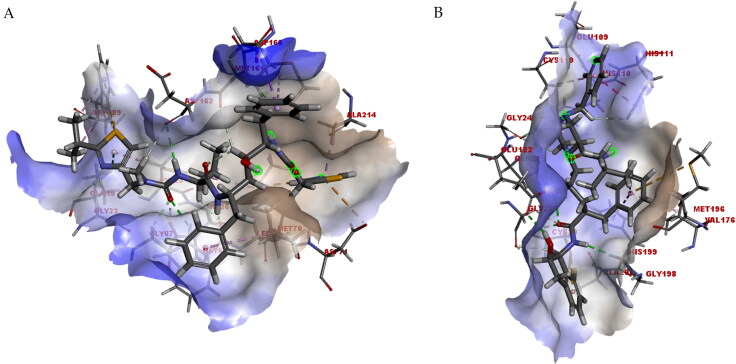
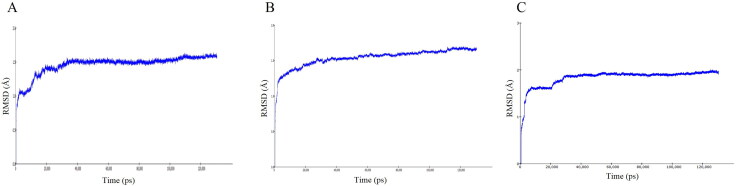
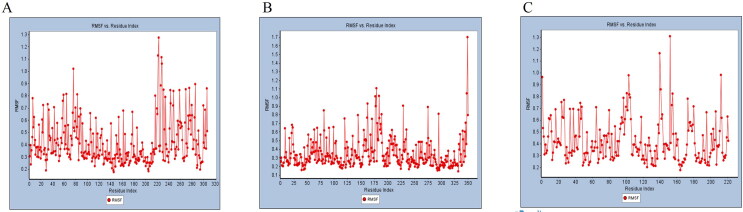
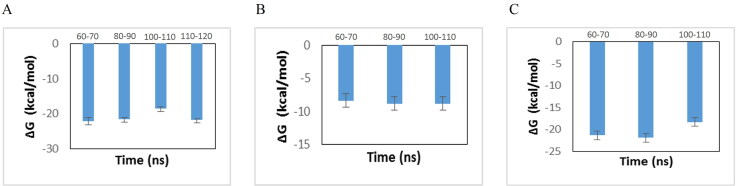
COVID-19, which has emerged recently as a pandemic viral infection caused by SARS-coronavirus 2 has spread rapidly around the world, creating a public health emergency. The current situation demands an effective therapeutic strategy to control the disease using drugs that are approved, or by inventing new ones. The present study examines the possible repurposing of existing anti-viral protease inhibitor drugs. For this, the structural features of the viral spike protein, the substrate for host cell protease and main protease of the available SARS CoV-2 isolates were established by comparing with related viruses for which antiviral drugs are effective. The results showed 97% sequence similarity among SARS and SARS-CoV-2 main protease and has same cleavage site positions and ACE2 receptor binding region as in the SARS-CoV spike protein. Though both are N-glycosylated, unlike SARS-CoV, human SARS-CoV-2 S-protein was O-glycosylated as well. Molecular docking studies were done to explore the role of FDA approved protease inhibitors to control SARS-CoV-2 replication. The results indicated that, Ritonavir has the highest potency to block SARS-CoV-2 main protease and human TMPRSS2, a host cell factor that aids viral infection. Other drugs such as Indinavir and Atazanavir also showed favourable binding with Cathepsin B/L that helped viral fusion with the host cell membrane. Further molecular dynamics simulation and MM-PBSA binding free energy calculations confirmed the stability of protein-drug complexes. These results suggest that protease inhibitors particularly Ritonavir, either alone or in combination with other drugs such as Atazanavir, have the potential to treat COVID 19.Communicated by Ramaswamy H. Sarma.
Keywords: COVID-19; TMPRSS2; main protease; molecular docking; spike protein.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Bagdonaite, I., Nordén, R., Joshi, H. J., Dabelsteen, S., Nyström, K., Vakhrushev, S. Y., Olofsson, S., & Wandall, H. H. (2015). A strategy for O-glycoproteomics of enveloped viruses-the O-glycoproteome of herpes simplex virus type 1. PLoS Pathogens, 11(4), e1004784. 10.1371/journal.ppat.1004784 - DOI - PMC - PubMed
-
- Cao, B., Wang, Y., Wen, D., Liu, W., Wang, J., Fan, G., Ruan, L., Song, B., Cai, Y., Wei, M., Li, X., Xia, J., Chen, N., Xiang, J., Yu, T., Bai, T., Xie, X., Zhang, L., Li, C., … Wang, C. (2020). A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. The New England Journal of Medicine, 382(19), 1787–1799. 10.1056/NEJMoa2001282 - DOI - PMC - PubMed
-
- Chan, K. S., Lai, S. T., Chu, C. M., Tsui, E., Tam, C. Y., & Wong, M. M. (2003). Treatment of severe acute respiratory syndrome with lopinavir/ritonavir: A multicentre retrospective matched cohort study. Hong Kong Medical Journal, 9(6), 399–406. - PubMed
-
- Chen, J., Lee, K. H., Steinhauer, D. A., Stevens, D. J., Skehel, J. J., & Wiley, D. C. (1998). Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell, 95(3), 409–417. 10.1016/S0092-8674(00)81771-7 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous