

Structural basis for the action of the drug trametinib at KSR-bound MEK
- PMID: 32927473
- PMCID: PMC7746607
- DOI: 10.1038/s41586-020-2760-4
Structural basis for the action of the drug trametinib at KSR-bound MEK
Abstract
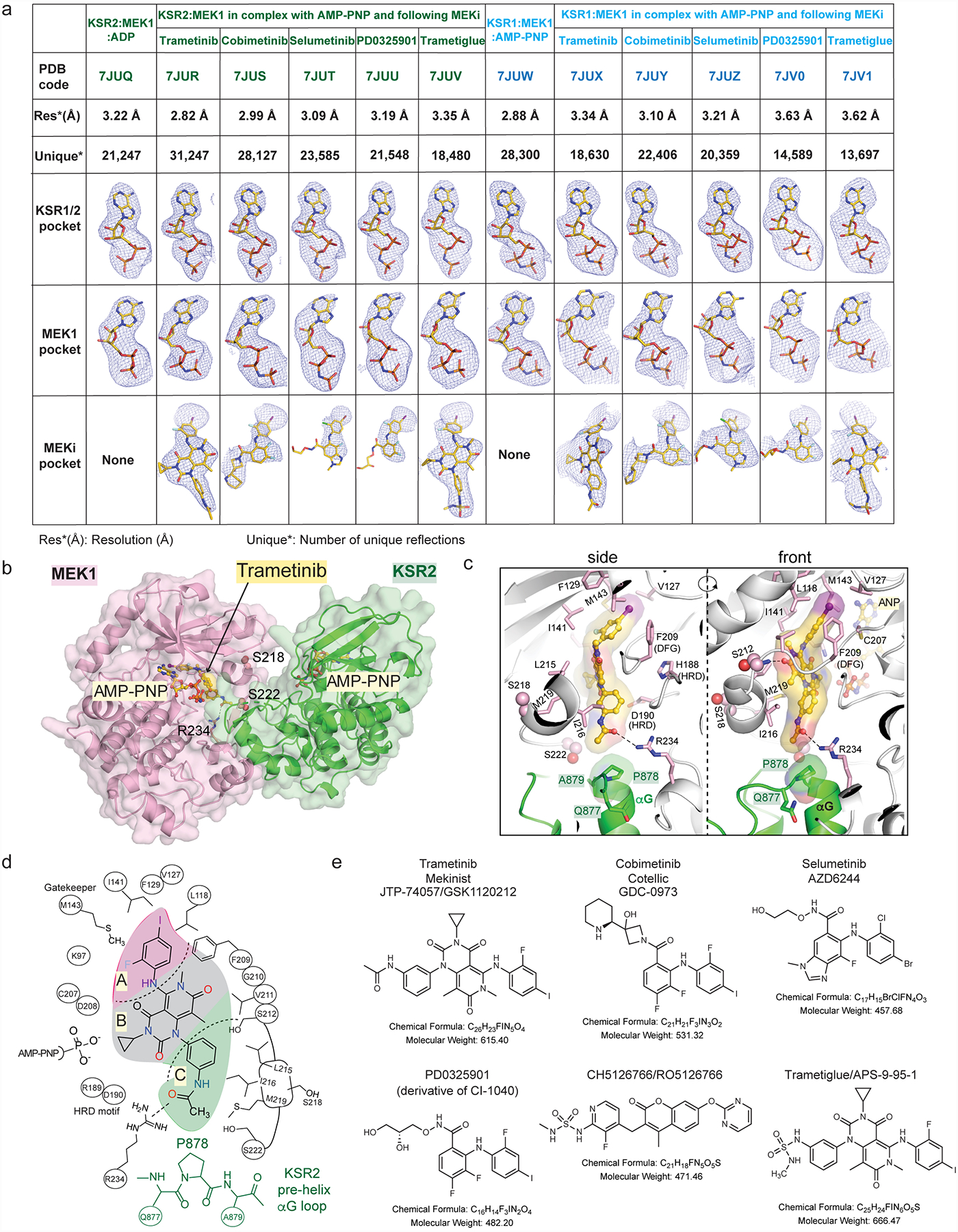
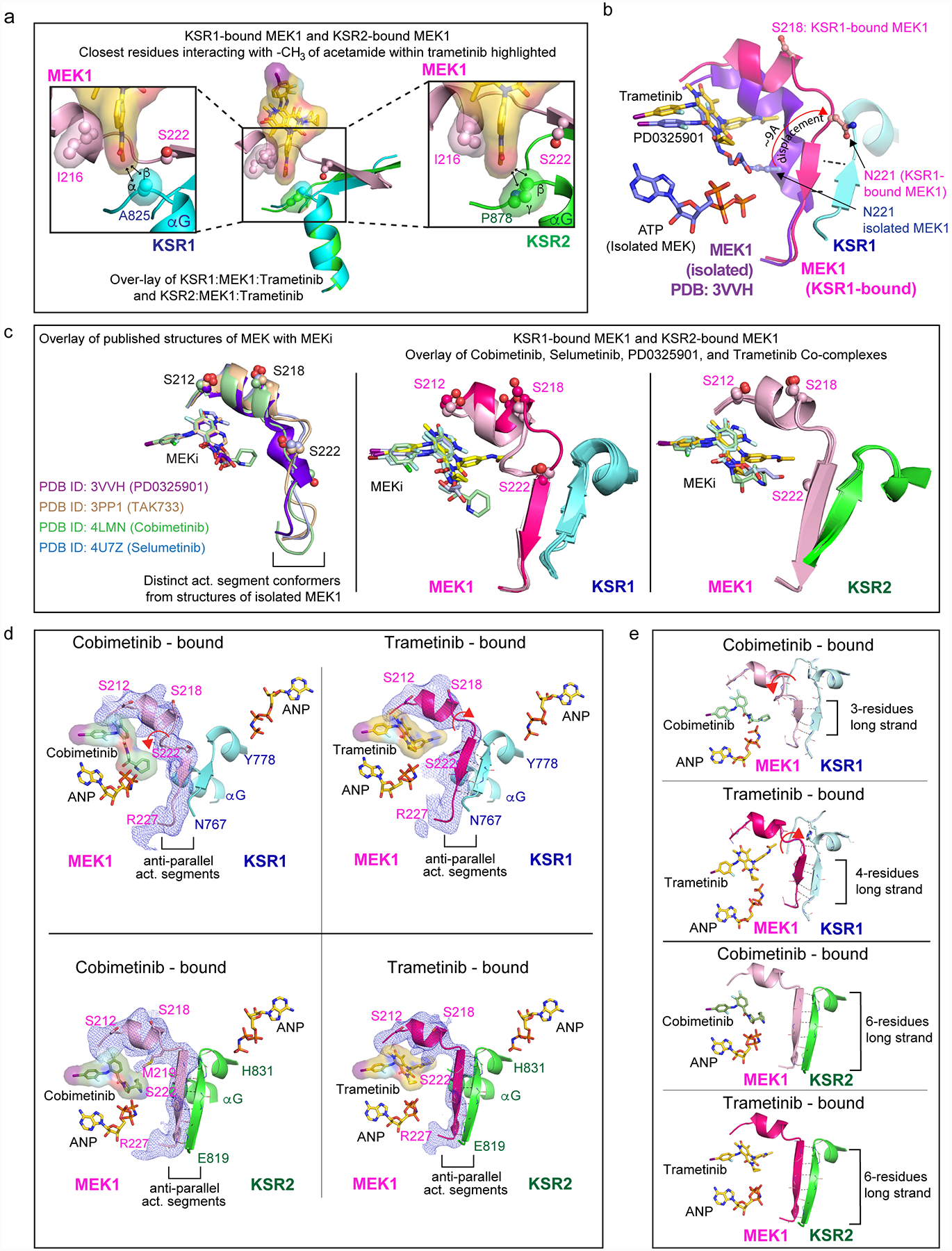
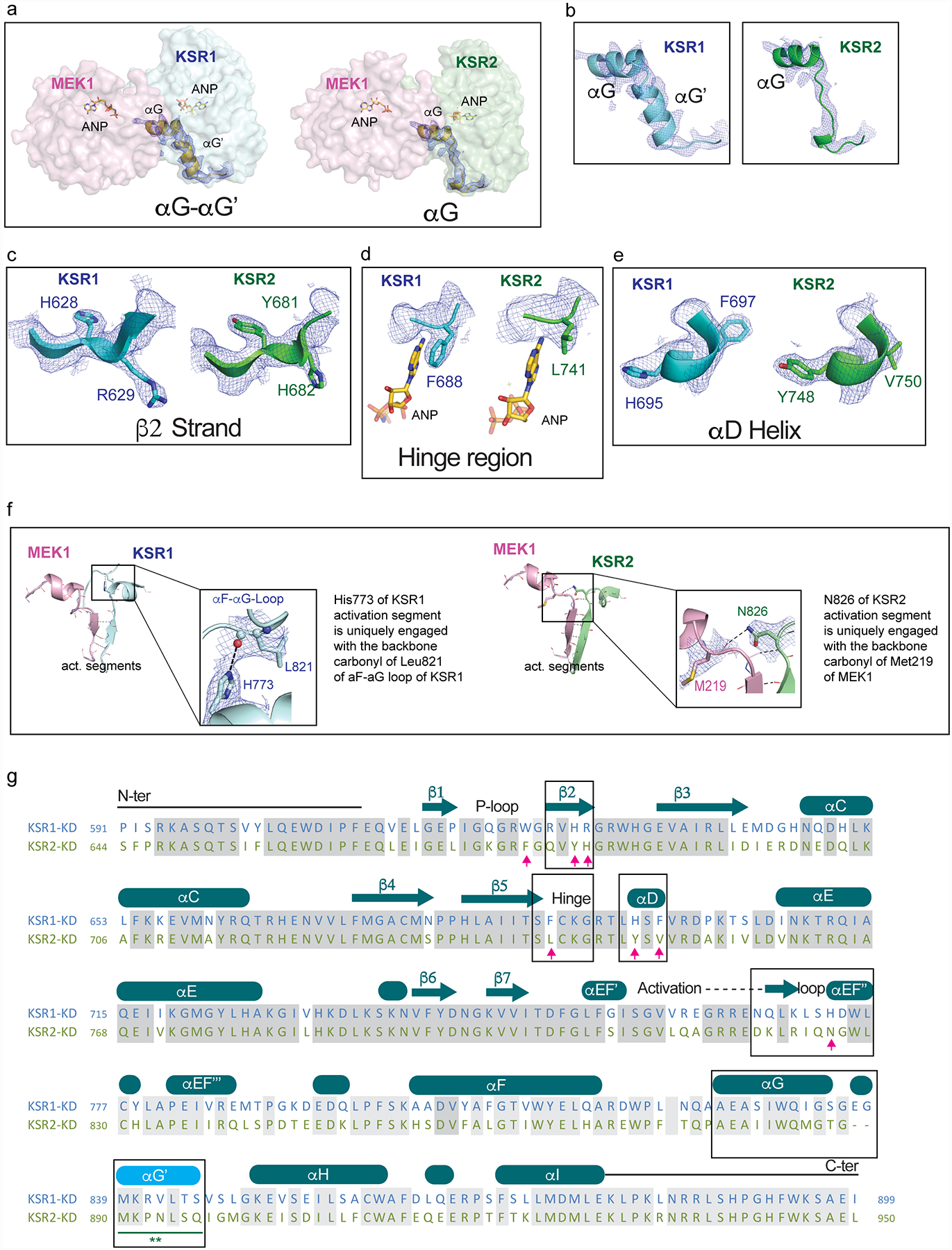
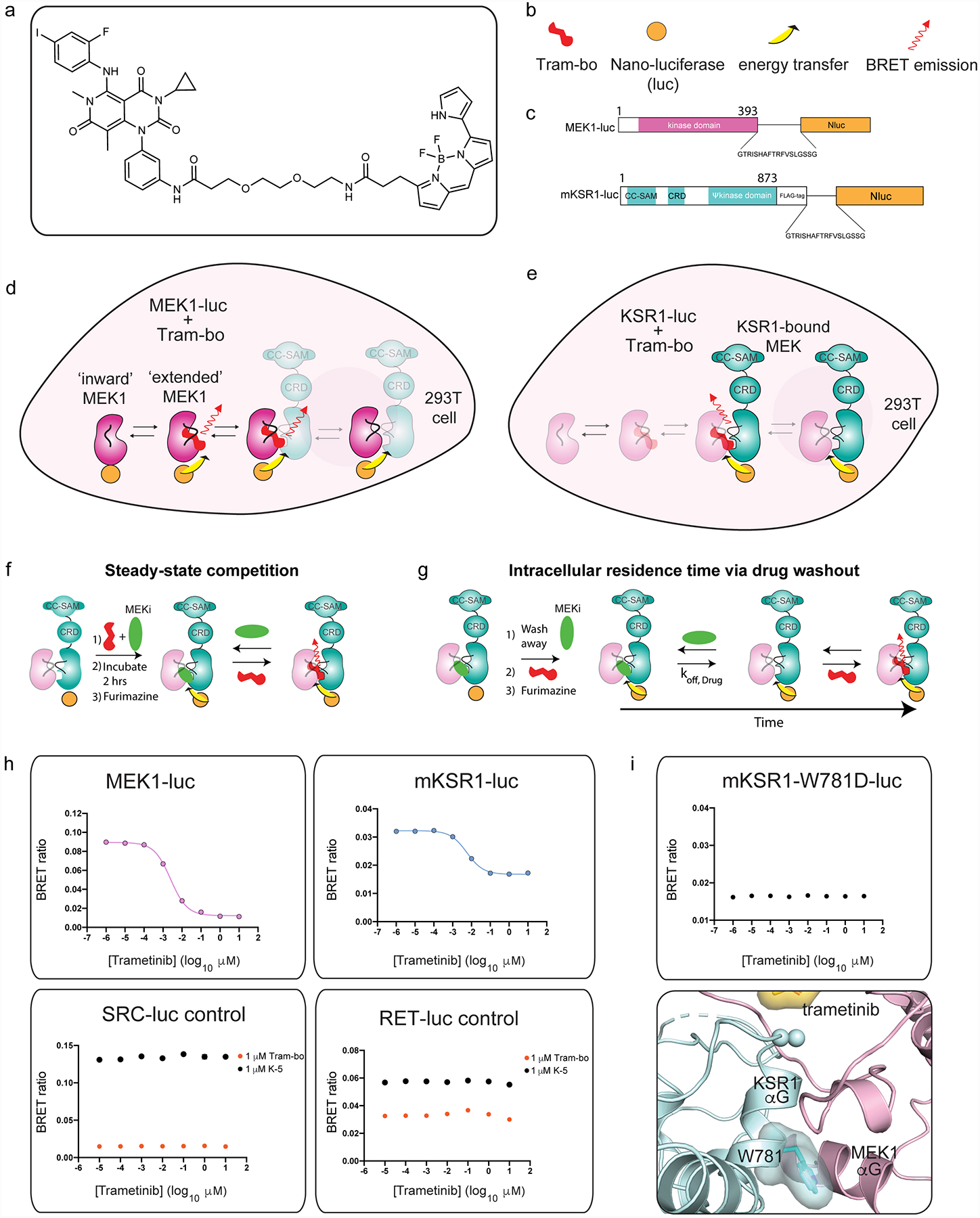
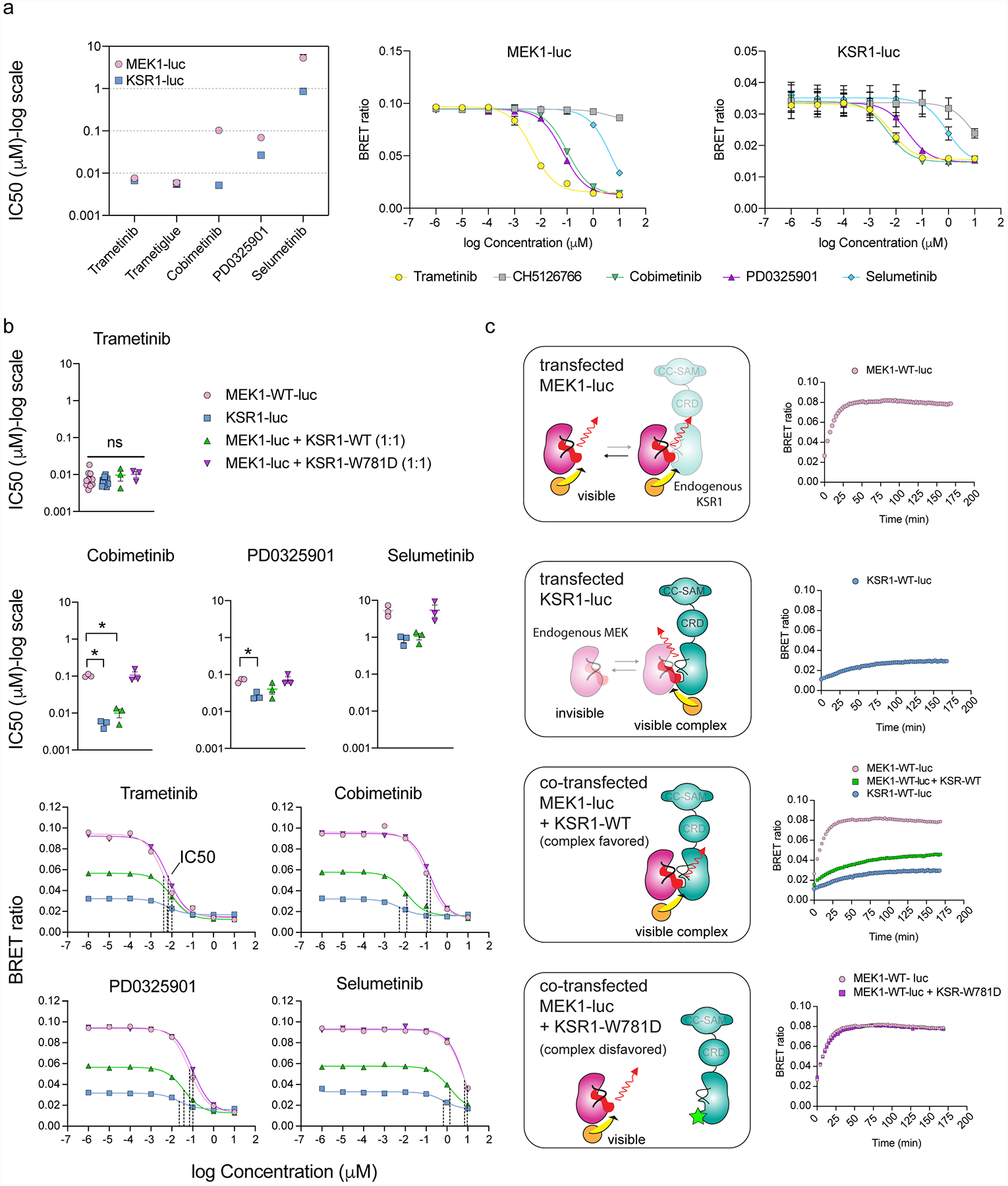
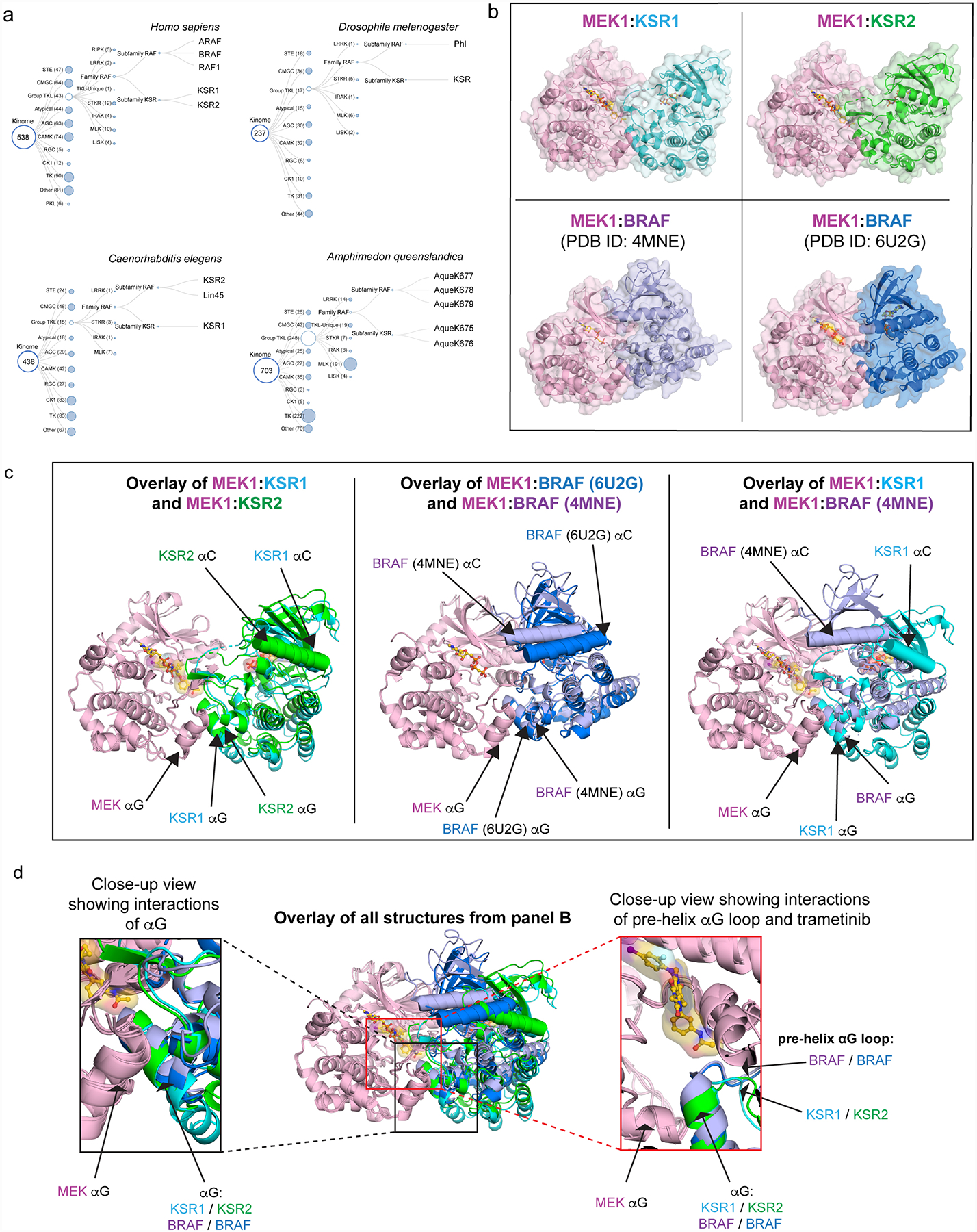
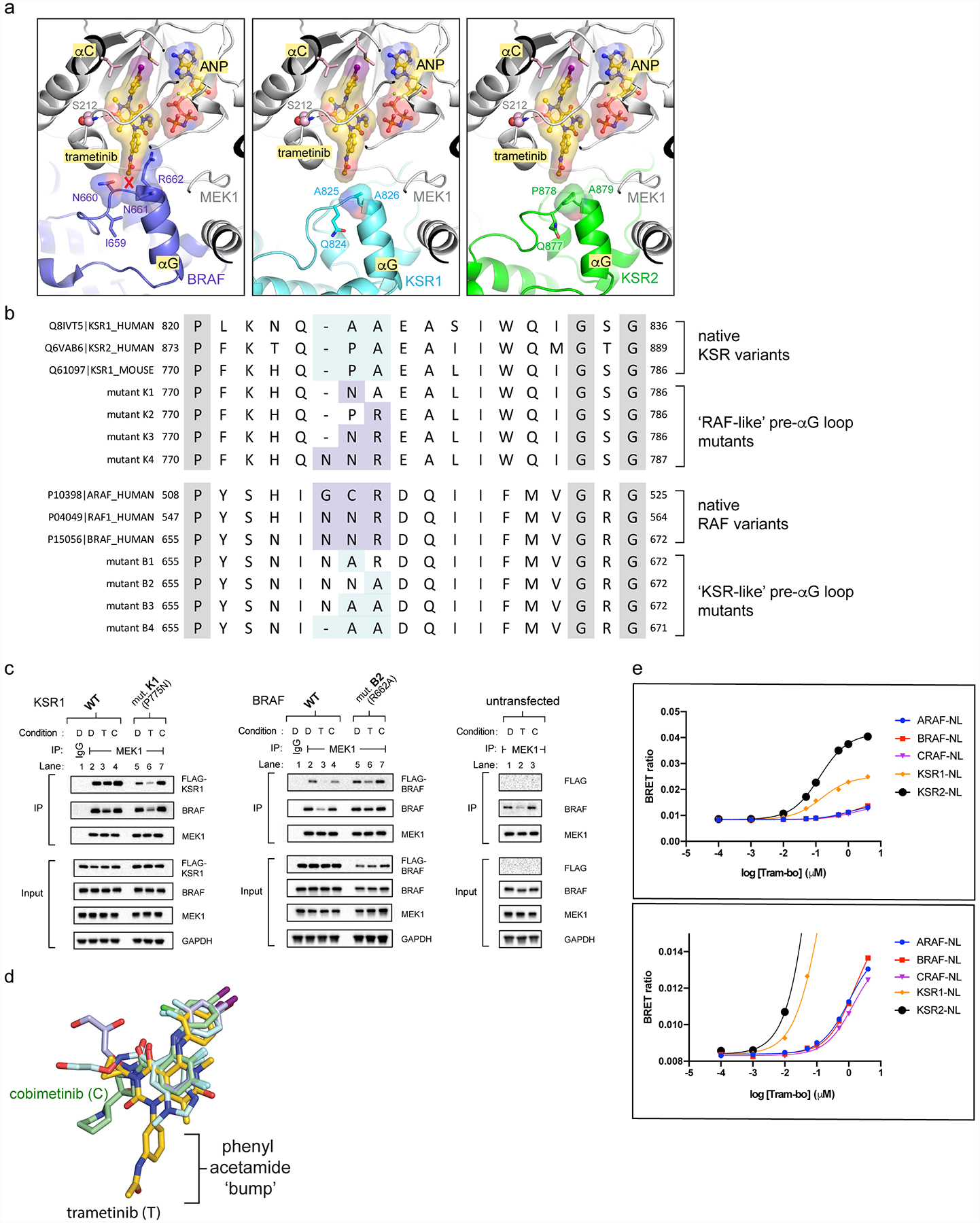
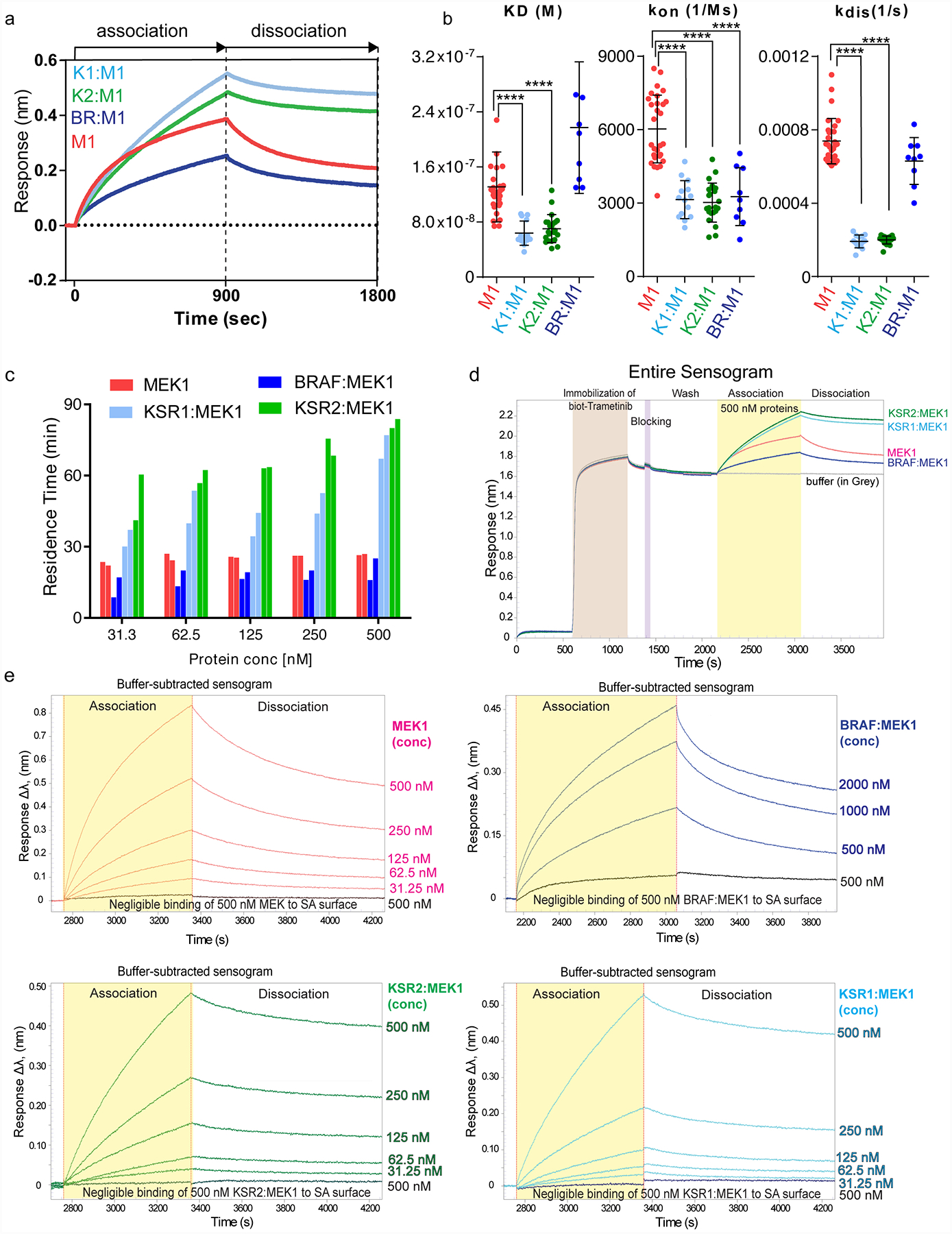
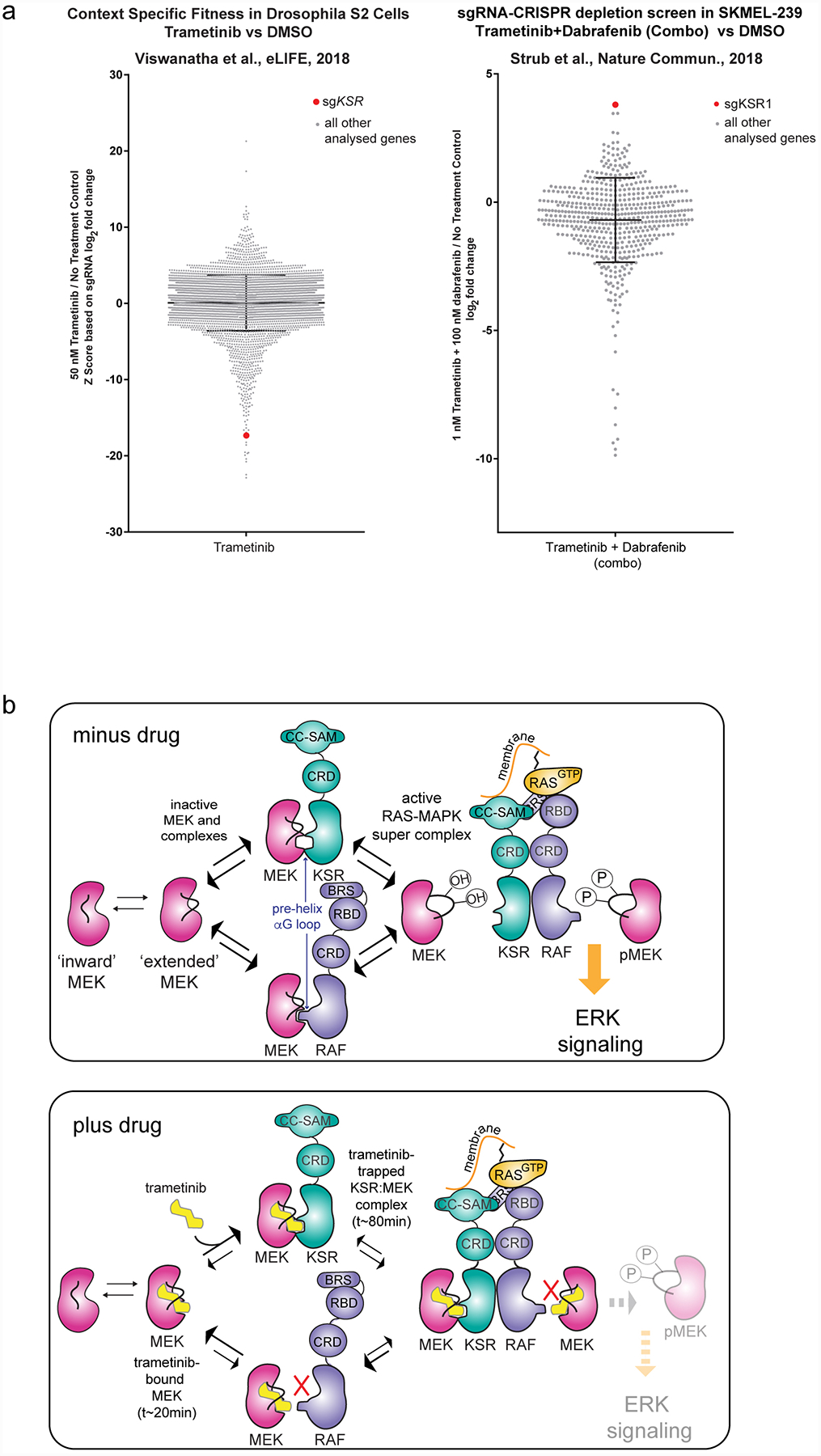
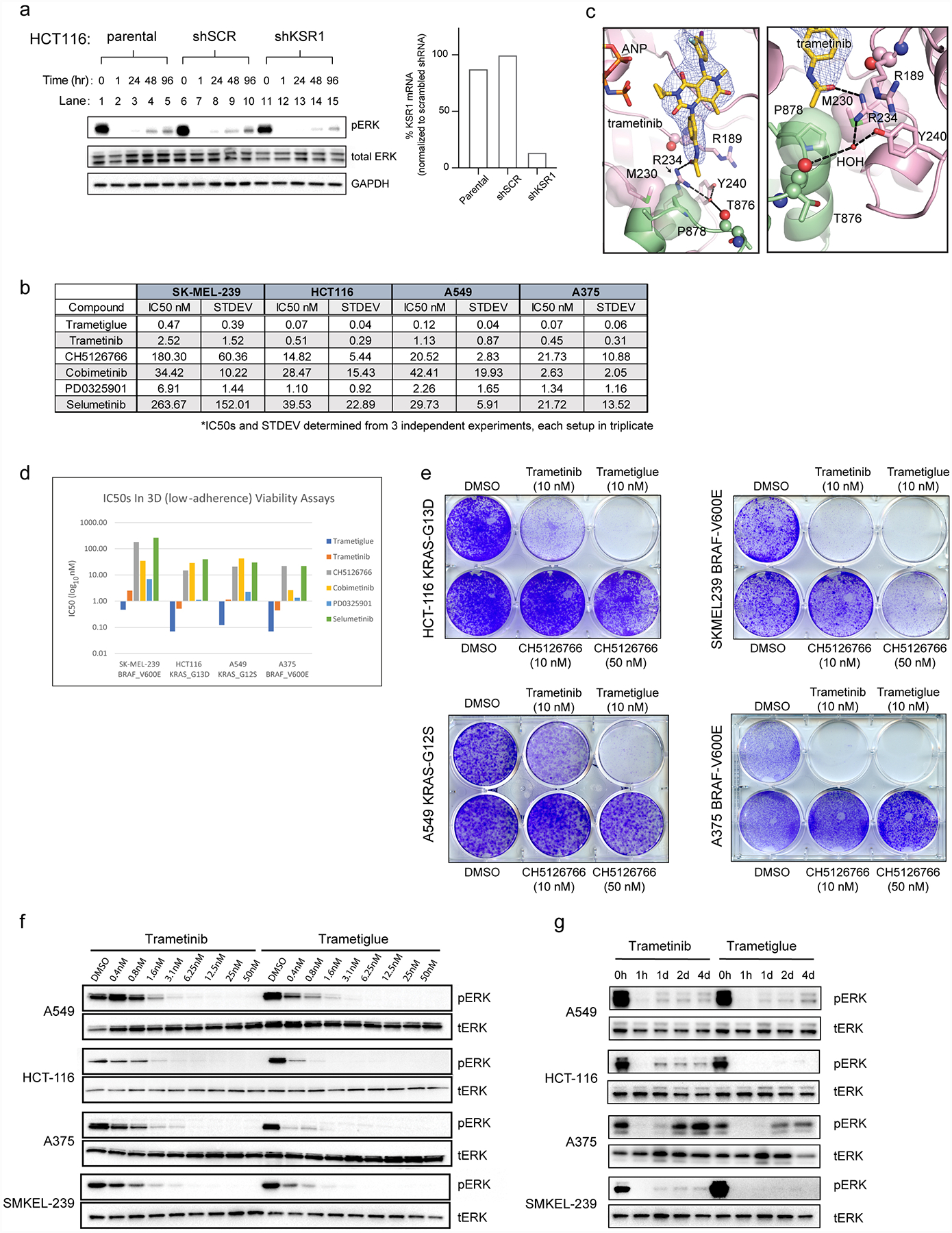
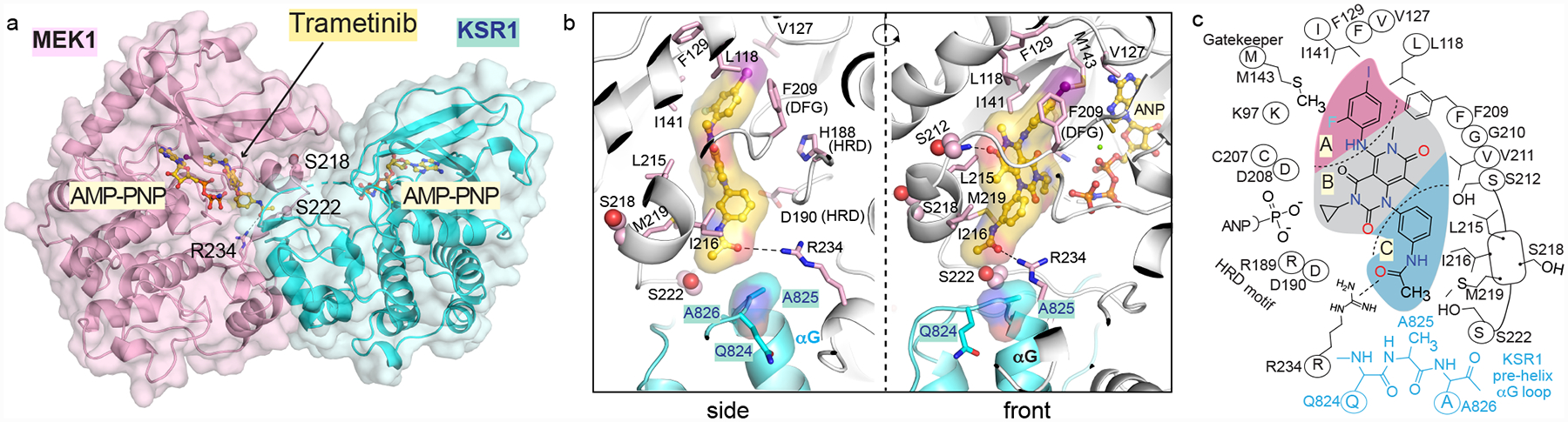
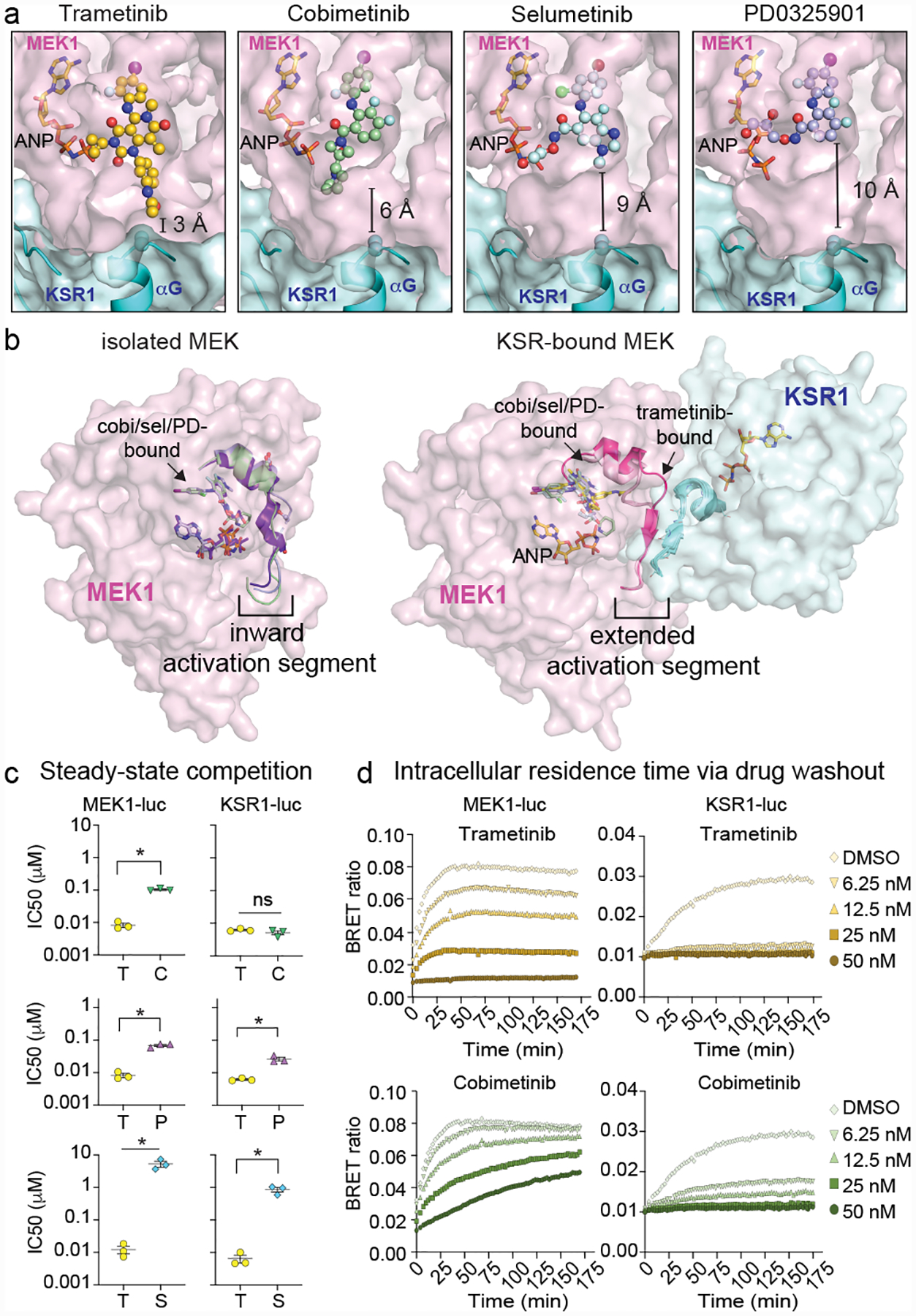
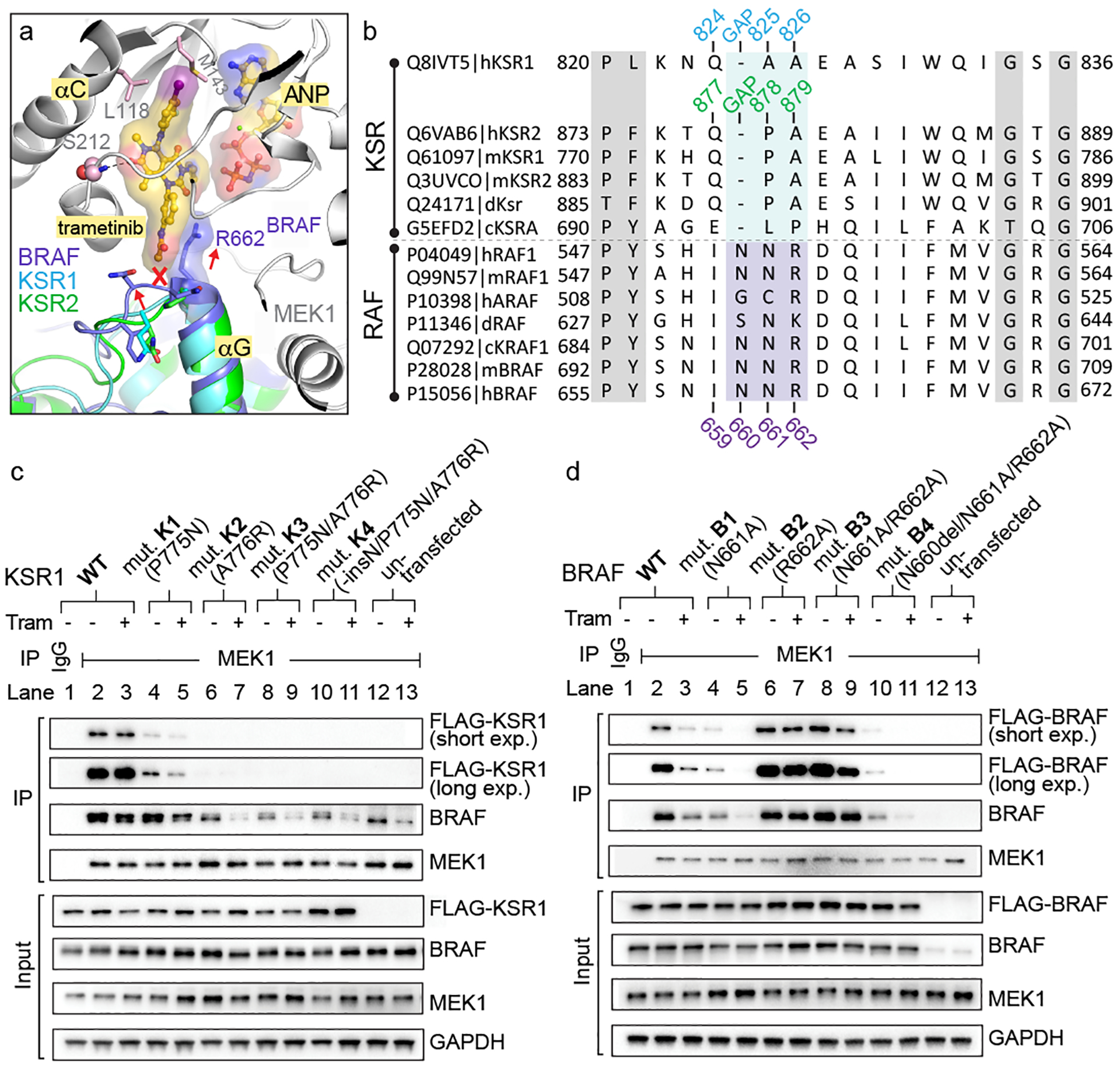
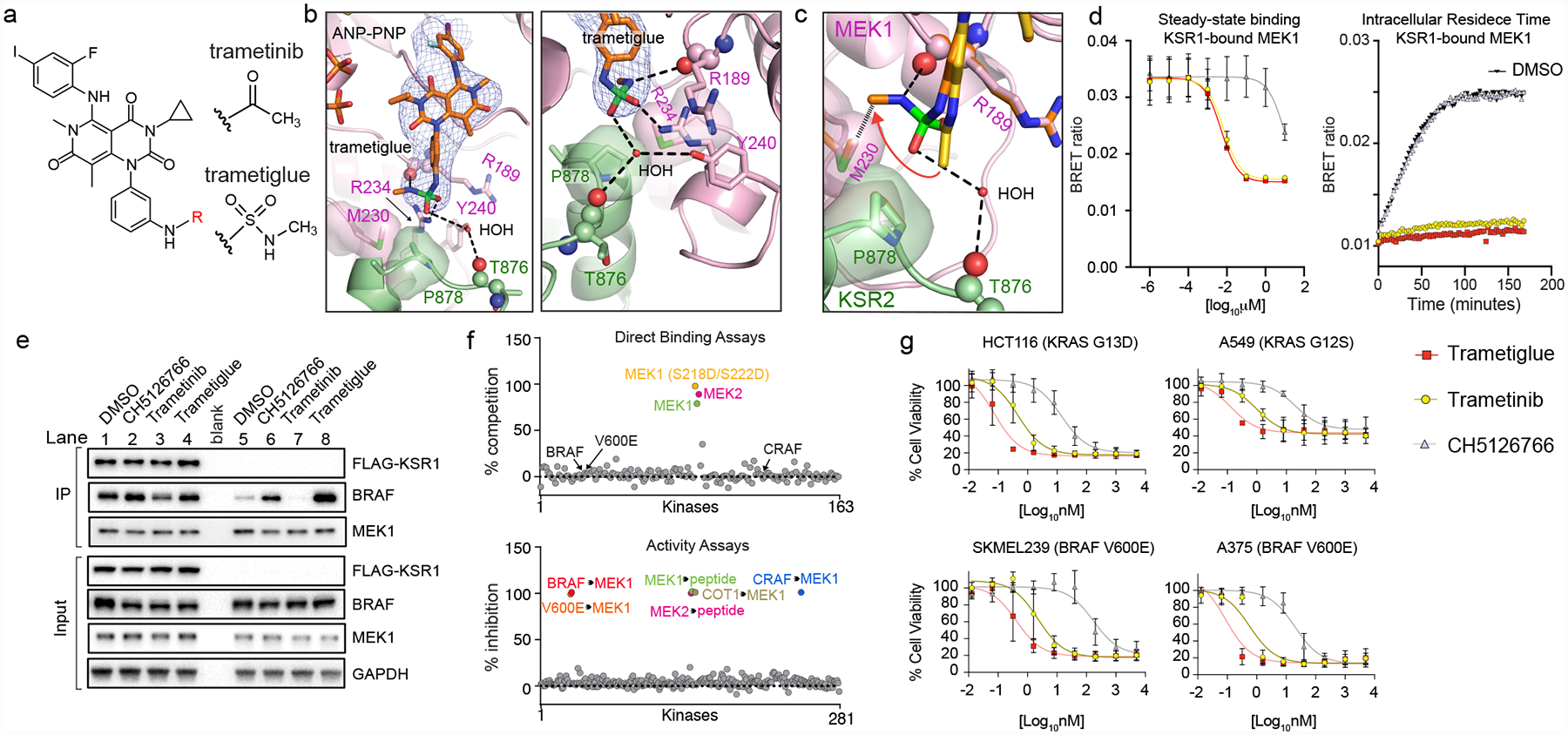
The MAPK/ERK kinase MEK is a shared effector of the frequent cancer drivers KRAS and BRAF that has long been pursued as a drug target in oncology1, and more recently in immunotherapy2,3 and ageing4. However, many MEK inhibitors are limited owing to on-target toxicities5-7 and drug resistance8-10. Accordingly, a molecular understanding of the structure and function of MEK within physiological complexes could provide a template for the design of safer and more effective therapies. Here we report X-ray crystal structures of MEK bound to the scaffold KSR (kinase suppressor of RAS) with various MEK inhibitors, including the clinical drug trametinib. The structures reveal an unexpected mode of binding in which trametinib directly engages KSR at the MEK interface. In the bound complex, KSR remodels the prototypical allosteric pocket of the MEK inhibitor, thereby affecting binding and kinetics, including the drug-residence time. Moreover, trametinib binds KSR-MEK but disrupts the related RAF-MEK complex through a mechanism that exploits evolutionarily conserved interface residues that distinguish these sub-complexes. On the basis of these insights, we created trametiglue, which limits adaptive resistance to MEK inhibition by enhancing interfacial binding. Our results reveal the plasticity of an interface pocket within MEK sub-complexes and have implications for the design of next-generation drugs that target the RAS pathway.
Conflict of interest statement
Figures
References
-
- Zhao Y & Adjei AA The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol 11, 385–400 (2014). - PubMed
-
- Ebert PJR et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 44, 609–621 (2016). - PubMed
-
- Liu L et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res 21, 1639–1651 (2015). - PubMed
-
- LoRusso PM et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin. Cancer Res 16, 1924–1937 (2010). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
