

Infection Dynamics of Swine Influenza Virus in a Danish Pig Herd Reveals Recurrent Infections with Different Variants of the H1N2 Swine Influenza A Virus Subtype
- PMID: 32927910
- PMCID: PMC7551734
- DOI: 10.3390/v12091013
Infection Dynamics of Swine Influenza Virus in a Danish Pig Herd Reveals Recurrent Infections with Different Variants of the H1N2 Swine Influenza A Virus Subtype
Abstract
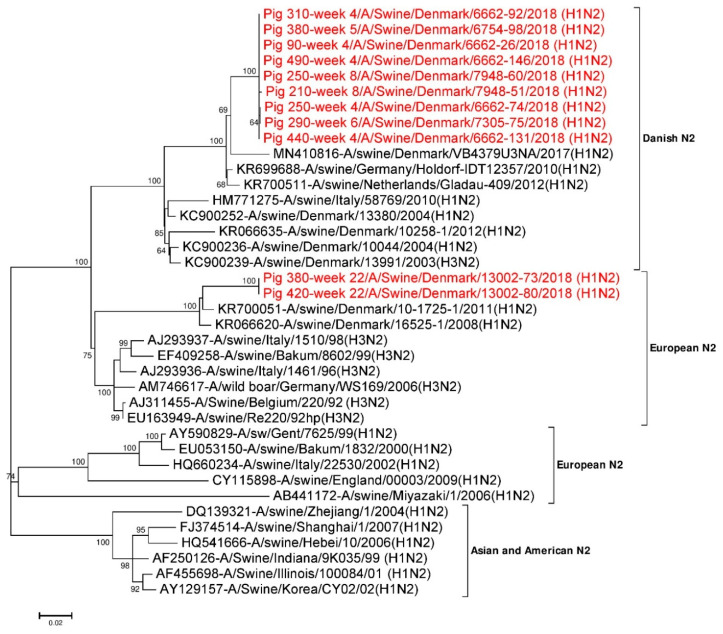
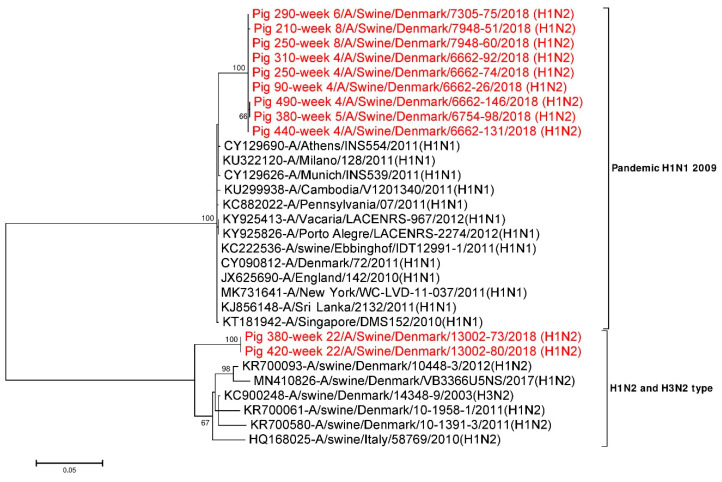
Influenza A virus (IAV) in swine, so-called swine influenza A virus (swIAV), causes respiratory illness in pigs around the globe. In Danish pig herds, a H1N2 subtype named H1N2dk is one of the main circulating swIAV. In this cohort study, the infection dynamic of swIAV was evaluated in a Danish pig herd by sampling and PCR testing of pigs from two weeks of age until slaughter at 22 weeks of age. In addition, next generation sequencing (NGS) was used to identify and characterize the complete genome of swIAV circulating in the herd, and to examine the antigenic variability in the antigenic sites of the virus hemagglutinin (HA) and neuraminidase (NA) proteins. Overall, 76.6% of the pigs became PCR positive for swIAV during the study, with the highest prevalence at four weeks of age. Detailed analysis of the virus sequences obtained showed that the majority of mutations occurred at antigenic sites in the HA and NA proteins of the virus. At least two different H1N2 variants were found to be circulating in the herd; one H1N2 variant was circulating at the sow and nursery sites, while another H1N2 variant was circulating at the finisher site. Furthermore, it was demonstrated that individual pigs had recurrent swIAV infections with the two different H1N2 variants, but re-infection with the same H1N2 variant was also observed. Better understandings of the epidemiology, genetic and antigenic diversity of swIAV may help to design better health interventions for the prevention and control of swIAV infections in the herds.
Keywords: H1N2; antigenic diversity; phylogenetic analysis; pig herd; prevalence; swine influenza A virus (swIAV).
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Figures

References
-
- Verhagen J.H., Munster V.J., Fouchier R.A. Genetics and Evolution of Infectious Disease. Elsevier; Amsterdam, The Netherlands: 2011. Ecology and evolution of avian influenza viruses; pp. 729–749.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
