

Cytotoxic CD8+ T cells in cancer and cancer immunotherapy
- PMID: 32929195
- PMCID: PMC7853123
- DOI: 10.1038/s41416-020-01048-4
Cytotoxic CD8+ T cells in cancer and cancer immunotherapy
Abstract
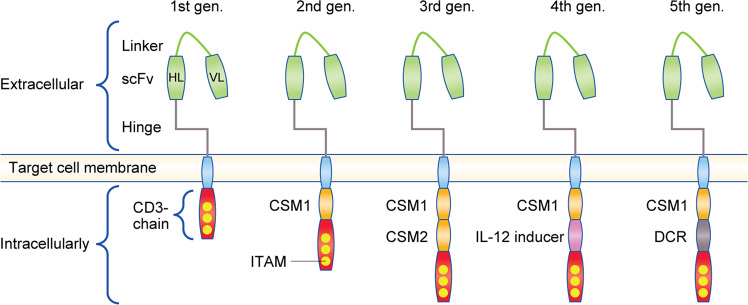
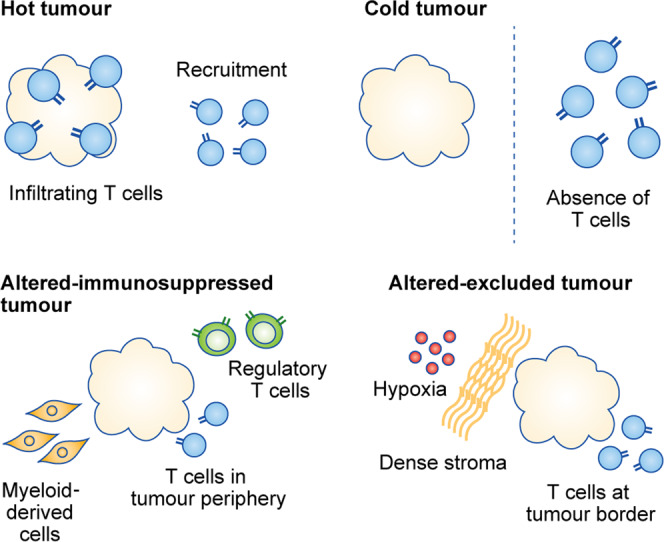
The functions of, and interactions between, the innate and adaptive immune systems are vital for anticancer immunity. Cytotoxic T cells expressing cell-surface CD8 are the most powerful effectors in the anticancer immune response and form the backbone of current successful cancer immunotherapies. Immune-checkpoint inhibitors are designed to target immune-inhibitory receptors that function to regulate the immune response, whereas adoptive cell-transfer therapies use CD8+ T cells with genetically modified receptors-chimaeric antigen receptors-to specify and enhance CD8+ T-cell functionality. New generations of cytotoxic T cells with genetically modified or synthetic receptors are being developed and evaluated in clinical trials. Furthermore, combinatory regimens might optimise treatment effects and reduce adverse events. This review summarises advances in research on the most prominent immune effectors in cancer and cancer immunotherapy, cytotoxic T cells, and discusses possible implications for future cancer treatment.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
