

Single-cell multiomics: technologies and data analysis methods
- PMID: 32929225
- PMCID: PMC8080692
- DOI: 10.1038/s12276-020-0420-2
Single-cell multiomics: technologies and data analysis methods
Abstract
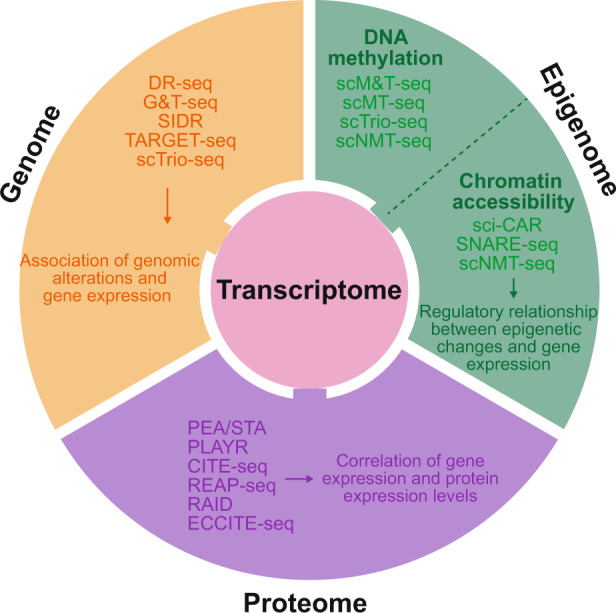
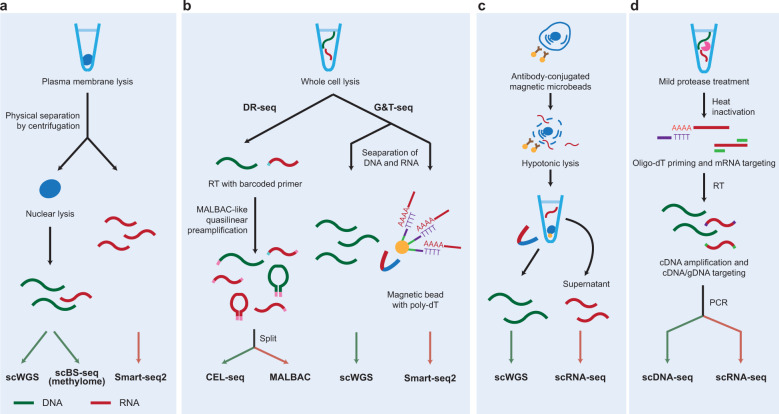
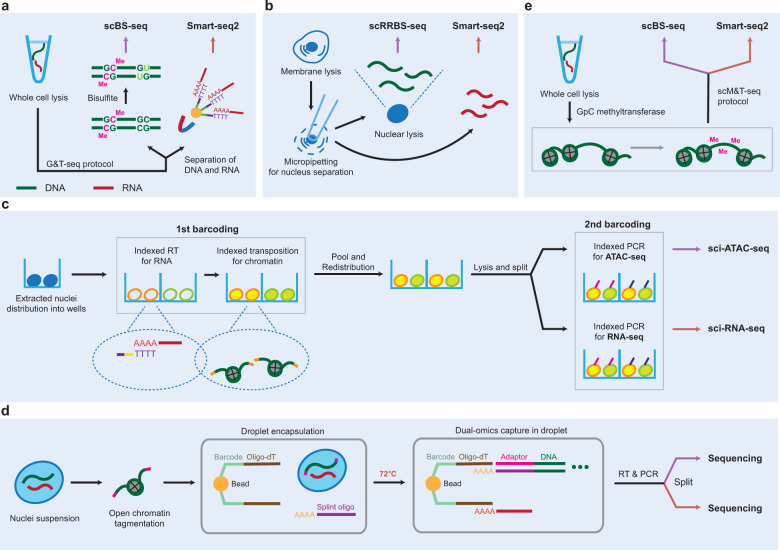
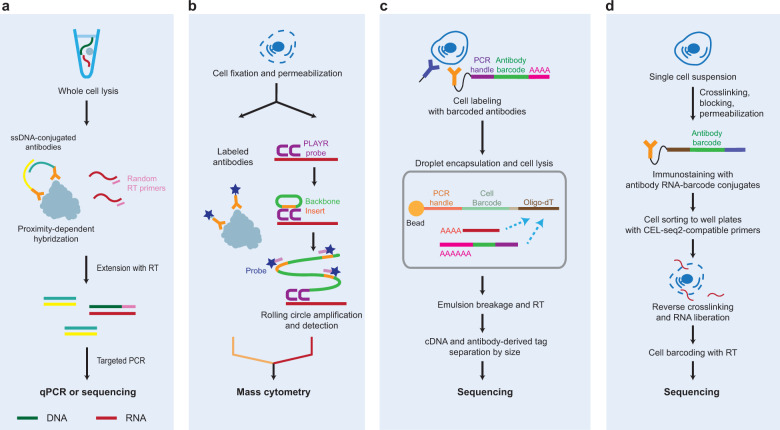
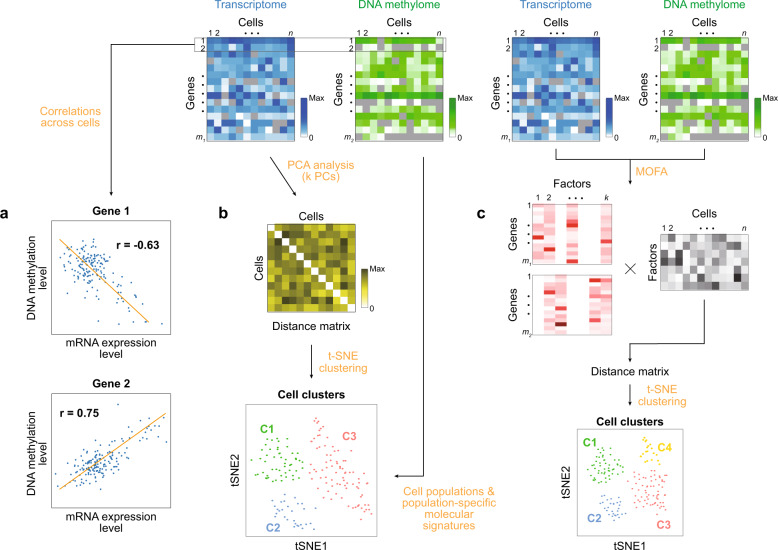
Advances in single-cell isolation and barcoding technologies offer unprecedented opportunities to profile DNA, mRNA, and proteins at a single-cell resolution. Recently, bulk multiomics analyses, such as multidimensional genomic and proteogenomic analyses, have proven beneficial for obtaining a comprehensive understanding of cellular events. This benefit has facilitated the development of single-cell multiomics analysis, which enables cell type-specific gene regulation to be examined. The cardinal features of single-cell multiomics analysis include (1) technologies for single-cell isolation, barcoding, and sequencing to measure multiple types of molecules from individual cells and (2) the integrative analysis of molecules to characterize cell types and their functions regarding pathophysiological processes based on molecular signatures. Here, we summarize the technologies for single-cell multiomics analyses (mRNA-genome, mRNA-DNA methylation, mRNA-chromatin accessibility, and mRNA-protein) as well as the methods for the integrative analysis of single-cell multiomics data.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
