

Single-cell transcriptomics in cancer: computational challenges and opportunities
- PMID: 32929226
- PMCID: PMC8080633
- DOI: 10.1038/s12276-020-0422-0
Single-cell transcriptomics in cancer: computational challenges and opportunities
Abstract
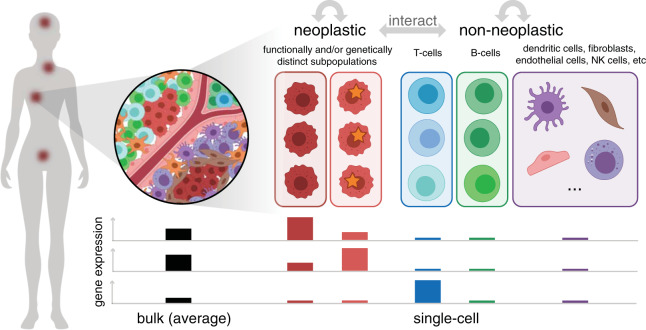
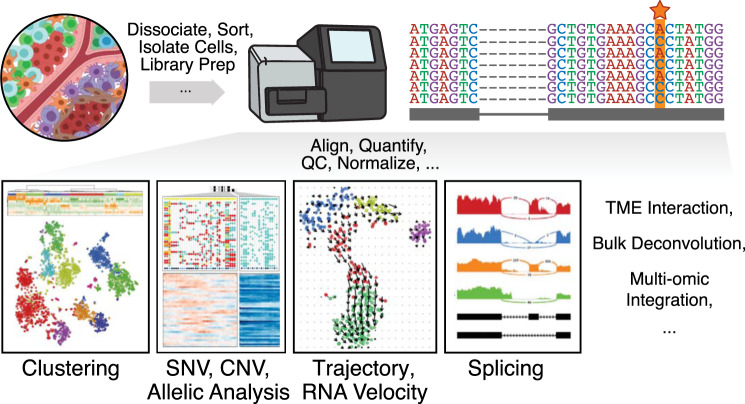
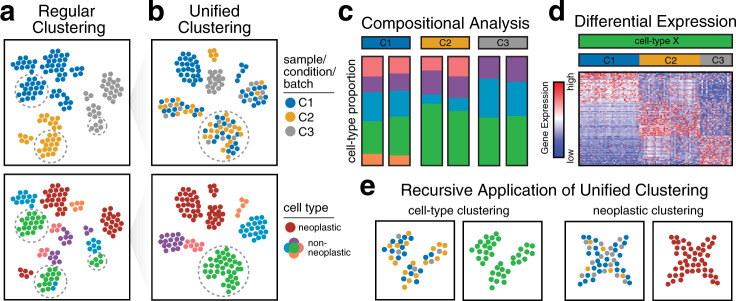
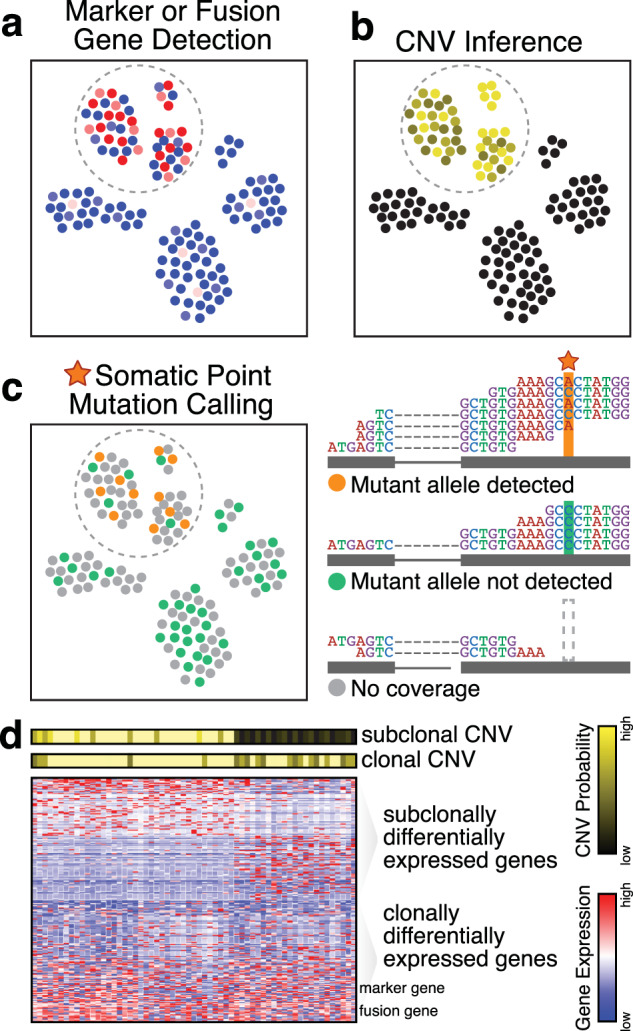
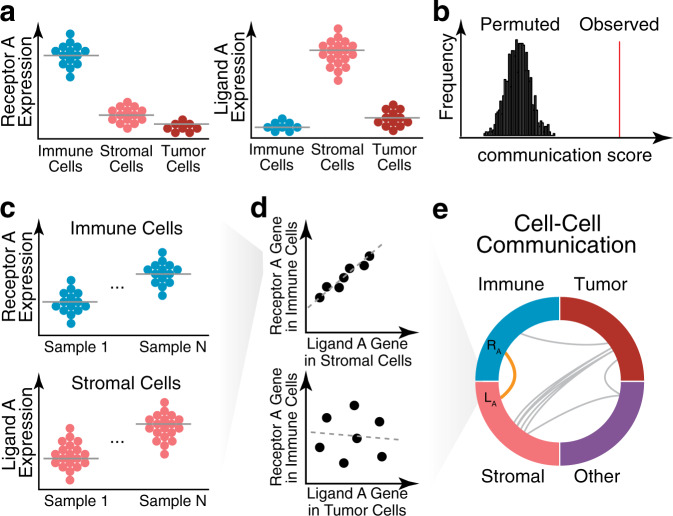
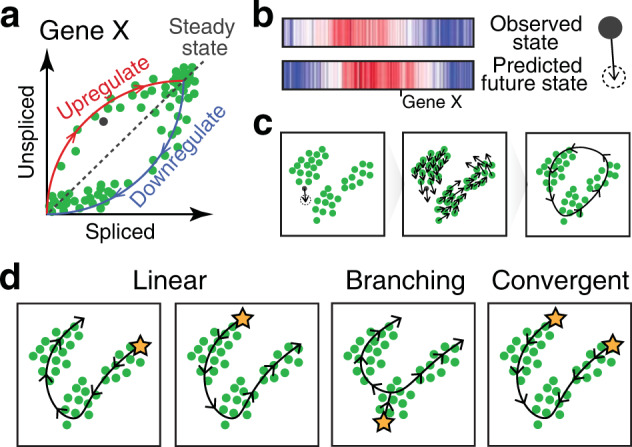
Intratumor heterogeneity is a common characteristic across diverse cancer types and presents challenges to current standards of treatment. Advancements in high-throughput sequencing and imaging technologies provide opportunities to identify and characterize these aspects of heterogeneity. Notably, transcriptomic profiling at a single-cell resolution enables quantitative measurements of the molecular activity that underlies the phenotypic diversity of cells within a tumor. Such high-dimensional data require computational analysis to extract relevant biological insights about the cell types and states that drive cancer development, pathogenesis, and clinical outcomes. In this review, we highlight emerging themes in the computational analysis of single-cell transcriptomics data and their applications to cancer research. We focus on downstream analytical challenges relevant to cancer research, including how to computationally perform unified analysis across many patients and disease states, distinguish neoplastic from nonneoplastic cells, infer communication with the tumor microenvironment, and delineate tumoral and microenvironmental evolution with trajectory and RNA velocity analysis. We include discussions of challenges and opportunities for future computational methodological advancements necessary to realize the translational potential of single-cell transcriptomic profiling in cancer.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat. Rev. Cancer. 2012;12:323–334. - PubMed
-
- Dagogo-Jack, I. & Shaw, A. T. Tumour heterogeneity and resistance to cancer therapies. 10.1038/nrclinonc.2017.166 (2018). - PubMed
-
- Cieślik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2018;19:93–109. - PubMed
-
- Suvà ML, Tirosh I. Single-cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol. Cell. 2019;75:7–12. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
