

Underlying features of epigenetic aging clocks in vivo and in vitro
- PMID: 32930491
- PMCID: PMC7576259
- DOI: 10.1111/acel.13229
Underlying features of epigenetic aging clocks in vivo and in vitro
Abstract
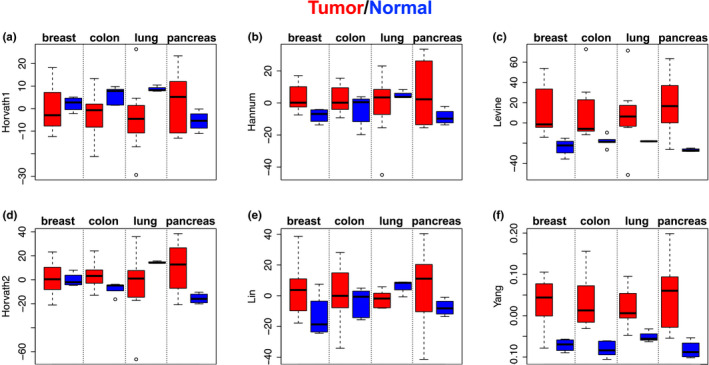
Epigenetic clocks, developed using DNA methylation data, have been widely used to quantify biological aging in multiple tissues/cells. However, many existing epigenetic clocks are weakly correlated with each other, suggesting they may capture different biological processes. We utilize multi-omics data from diverse human tissue/cells to identify shared features across eleven existing epigenetic clocks. Despite the striking lack of overlap in CpGs, multi-omics analysis suggested five clocks (Horvath1, Horvath2, Levine, Hannum, and Lin) share transcriptional associations conserved across purified CD14+ monocytes and dorsolateral prefrontal cortex. The pathways enriched in the shared transcriptional association suggested links between epigenetic aging and metabolism, immunity, and autophagy. Results from in vitro experiments showed that two clocks (Levine and Lin) were accelerated in accordance with two hallmarks of aging-cellular senescence and mitochondrial dysfunction. Finally, using multi-tissue data to deconstruct the epigenetic clock signals, we developed a meta-clock that demonstrated improved prediction for mortality and robustly related to hallmarks of aging in vitro than single clocks.
Keywords: DNA methylation; biological aging; cellular senescence; epigenetic clock; mitochondria.
© 2020 The Authors. Aging Cell published by Anatomical Society and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Ambatipudi, S. , Horvath, S. , Perrier, F. , Cuenin, C. , Hernandez‐Vargas, H. , Le Calvez‐Kelm, F. , … Herceg, Z. (2020). DNA methylome analysis identifies accelerated epigenetic ageing associated with postmenopausal breast cancer susceptibility. European Journal of Cancer, 75, 299–307. 10.1016/j.ejca.2017.01.014 - DOI - PMC - PubMed
-
- Florath, I. , Butterbach, K. , Muller, H. , Bewerunge‐Hudler, M. , & Brenner, H. (2014). Cross‐sectional and longitudinal changes in DNA methylation with age: an epigenome‐wide analysis revealing over 60 novel age‐associated CpG sites. Human Molecular Genetics, 23(5), 1186–1201. 10.1093/hmg/ddt531 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- U01 AG046152/AG/NIA NIH HHS/United States
- R01 AG042210/AG/NIA NIH HHS/United States
- R01 AG065403/AG/NIA NIH HHS/United States
- R01 NS078009/NS/NINDS NIH HHS/United States
- R01 AG039478/AG/NIA NIH HHS/United States
- U18 NS082140/NS/NINDS NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- R01 AG057911/AG/NIA NIH HHS/United States
- P30 AG021342/AG/NIA NIH HHS/United States
- U24 AG041689/AG/NIA NIH HHS/United States
- R00 AG056599/AG/NIA NIH HHS/United States
- R01 AG034374/AG/NIA NIH HHS/United States
- R01 AG057912/AG/NIA NIH HHS/United States
- R00 AG052604/AG/NIA NIH HHS/United States
- U01 AG016976/AG/NIA NIH HHS/United States
- P30 AG066508/AG/NIA NIH HHS/United States
- R01 AG036042/AG/NIA NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- R01 AG034504/AG/NIA NIH HHS/United States
- K99 AG056599/AG/NIA NIH HHS/United States
- R01 AG041232/AG/NIA NIH HHS/United States
- R01 AG036836/AG/NIA NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
