

Phenotypic diversity in ALS and the role of poly-conformational protein misfolding
- PMID: 32930869
- PMCID: PMC7956917
- DOI: 10.1007/s00401-020-02222-x
Phenotypic diversity in ALS and the role of poly-conformational protein misfolding
Abstract
In many types of familial amyotrophic lateral sclerosis (fALS), mutations cause proteins to gain toxic properties that mediate neurodegenerative processes. It is becoming increasingly clear that the proteins involved in ALS, and those responsible for a host of other neurodegenerative diseases, share many characteristics with a growing number of prion diseases. ALS is a heterogenous disease in which the majority of cases are sporadic in their etiology. Studies investigating the inherited forms of the disease are now beginning to provide evidence that some of this heterogeneity may be due to the existence of distinct conformations that ALS-linked proteins can adopt to produce the equivalent of prion strains. In this review, we discuss the in vitro and in vivo evidence that has been generated to better understand the characteristics of these proteins and how their tertiary structure may impact the disease phenotype.
Keywords: Amyotrophic lateral sclerosis; Prion; Strains; Superoxide dismutase-1; TDP-43.
Figures
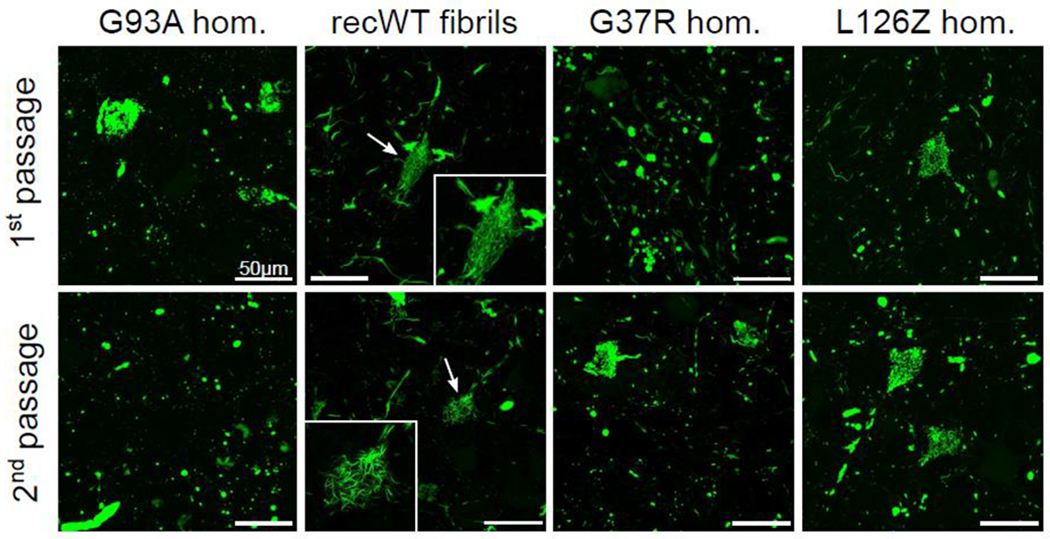
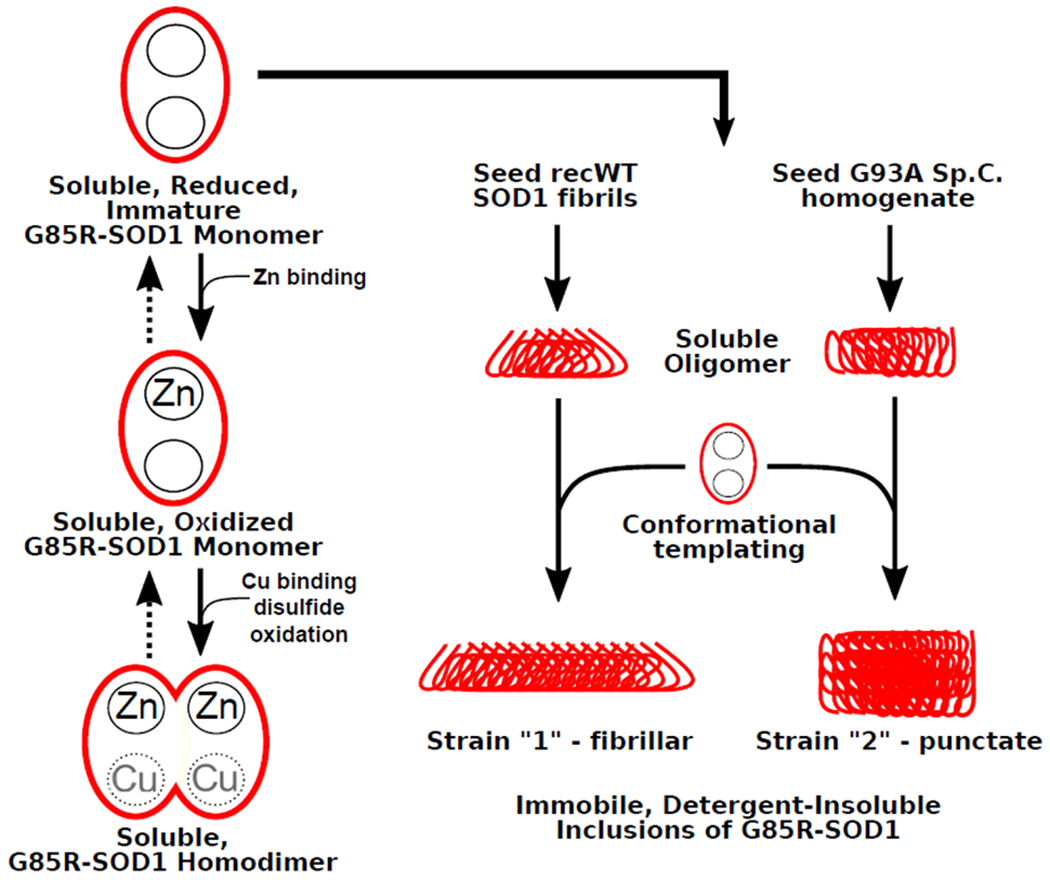
Similar articles
-
Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form.Acta Neuropathol. 2010 Mar;119(3):335-44. doi: 10.1007/s00401-010-0646-5. Epub 2010 Jan 29. Acta Neuropathol. 2010. PMID: 20111867
-
Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS.Acta Neuropathol. 2016 Dec;132(6):827-840. doi: 10.1007/s00401-016-1623-4. Epub 2016 Oct 4. Acta Neuropathol. 2016. PMID: 27704280 Free PMC article.
-
Prion-like mechanisms in amyotrophic lateral sclerosis.Handb Clin Neurol. 2018;153:337-354. doi: 10.1016/B978-0-444-63945-5.00018-0. Handb Clin Neurol. 2018. PMID: 29887144 Review.
-
The relevance of contact-independent cell-to-cell transfer of TDP-43 and SOD1 in amyotrophic lateral sclerosis.Biochim Biophys Acta Mol Basis Dis. 2017 Nov;1863(11):2762-2771. doi: 10.1016/j.bbadis.2017.07.007. Epub 2017 Jul 13. Biochim Biophys Acta Mol Basis Dis. 2017. PMID: 28711596 Free PMC article. Review.
-
Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations.Ann Neurol. 2007 May;61(5):427-34. doi: 10.1002/ana.21147. Ann Neurol. 2007. PMID: 17469116
Cited by
-
Peripheral administration of SOD1 aggregates does not transmit pathogenic aggregation to the CNS of SOD1 transgenic mice.Acta Neuropathol Commun. 2021 Jun 22;9(1):111. doi: 10.1186/s40478-021-01211-9. Acta Neuropathol Commun. 2021. PMID: 34158126 Free PMC article.
-
Amyloid fibril structures and ferroptosis activation induced by ALS-causing SOD1 mutations.Sci Adv. 2024 Nov;10(44):eado8499. doi: 10.1126/sciadv.ado8499. Epub 2024 Oct 30. Sci Adv. 2024. PMID: 39475611 Free PMC article.
-
Identification of Molecular Correlations Between DHRS4 and Progressive Neurodegeneration in Amyotrophic Lateral Sclerosis By Gene Co-Expression Network Analysis.Front Immunol. 2022 Apr 11;13:874978. doi: 10.3389/fimmu.2022.874978. eCollection 2022. Front Immunol. 2022. PMID: 35479082 Free PMC article.
-
Cryo-EM structure of an amyloid fibril formed by full-length human SOD1 reveals its conformational conversion.Nat Commun. 2022 Jun 17;13(1):3491. doi: 10.1038/s41467-022-31240-4. Nat Commun. 2022. PMID: 35715417 Free PMC article.
-
Protein Structure-based FUS Mutational Subtypes Are Associated With Protein Mislocalization in Amyotrophic Lateral Sclerosis Patients.Mol Neurobiol. 2025 May 24. doi: 10.1007/s12035-025-05085-z. Online ahead of print. Mol Neurobiol. 2025. PMID: 40413303
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
