

ALS Genetics: Gains, Losses, and Implications for Future Therapies
- PMID: 32931756
- PMCID: PMC7736125
- DOI: 10.1016/j.neuron.2020.08.022
ALS Genetics: Gains, Losses, and Implications for Future Therapies
Abstract
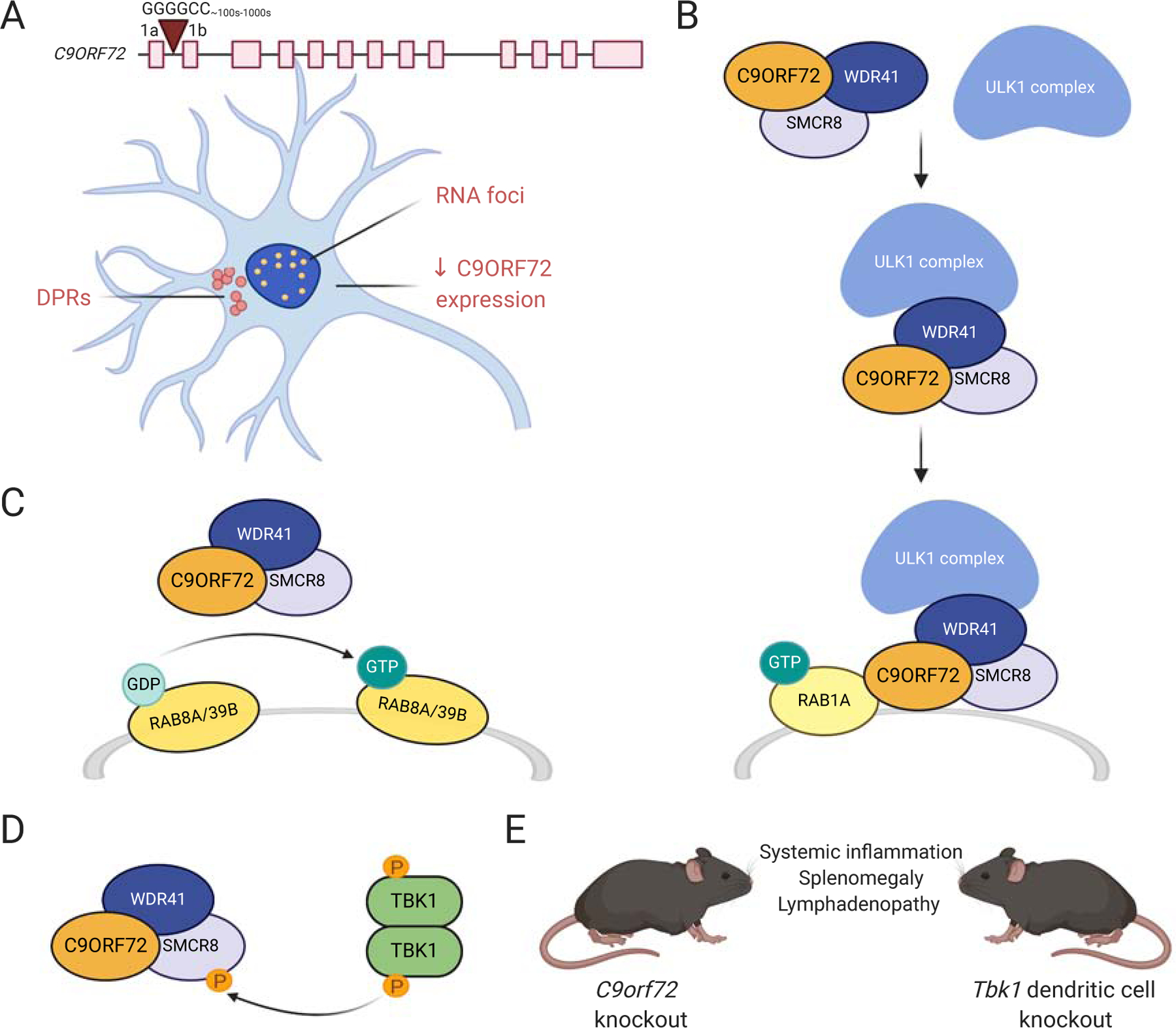
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder caused by the loss of motor neurons from the brain and spinal cord. The ALS community has made remarkable strides over three decades by identifying novel familial mutations, generating animal models, elucidating molecular mechanisms, and ultimately developing promising new therapeutic approaches. Some of these approaches reduce the expression of mutant genes and are in human clinical trials, highlighting the need to carefully consider the normal functions of these genes and potential contribution of gene loss-of-function to ALS. Here, we highlight known loss-of-function mechanisms underlying ALS, potential consequences of lowering levels of gene products, and the need to consider both gain and loss of function to develop safe and effective therapeutic strategies.
Keywords: ALS; C9ORF72; FUS; OPTN; SOD1; TARDBP; TBK1; TDP-43; gain of function; loss of function.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests A.D.G. has served as a consultant for Aquinnah Pharmaceuticals, Prevail Therapeutics, and Third Rock Ventures and is a scientific founder of Maze Therapeutics.
Figures




References
-
- Abo-Rady M, Kalmbach N, Pal A, Schludi C, Janosch A, Richter T, Freitag P, Bickle M, Kahlert A-K, Petri S, Stefanov S, Glass H, Staege S, Just W, Bhatnagar R, Edbauer D, Hermann A, Wegner F, Sterneckert JL, 2020. Knocking out C9ORF72 Exacerbates Axonal Trafficking Defects Associated with Hexanucleotide Repeat Expansion and Reduces Levels of Heat Shock Proteins. Stem Cell Rep. 14, 390–405. 10.1016/j.stemcr.2020.01.010 - DOI - PMC - PubMed
-
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, Bilican B, Chaum E, Chandran S, Shaw CE, Eggan KC, Maniatis T, Taylor JP, 2014. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. 10.1016/j.neuron.2013.12.018 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
