

Mouse Ataxin-2 Expansion Downregulates CamKII and Other Calcium Signaling Factors, Impairing Granule-Purkinje Neuron Synaptic Strength
- PMID: 32932600
- PMCID: PMC7555182
- DOI: 10.3390/ijms21186673
Mouse Ataxin-2 Expansion Downregulates CamKII and Other Calcium Signaling Factors, Impairing Granule-Purkinje Neuron Synaptic Strength
Abstract
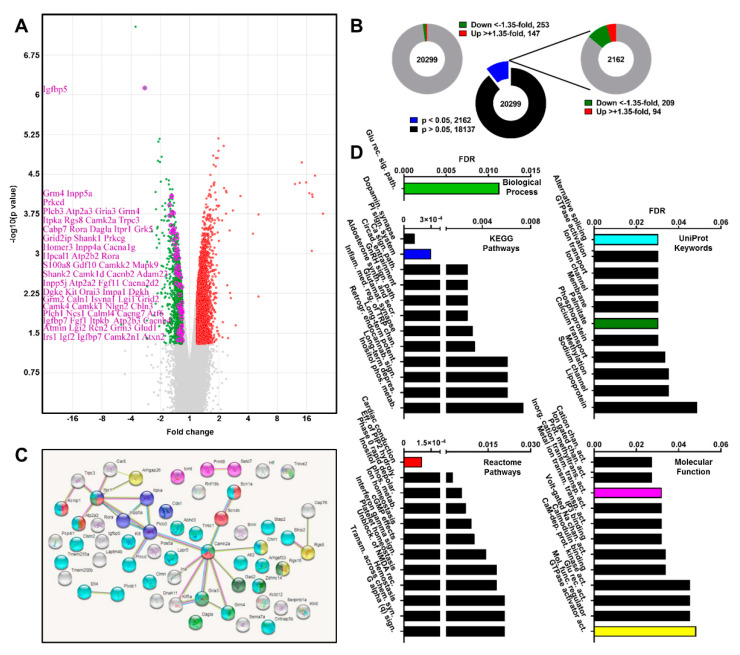
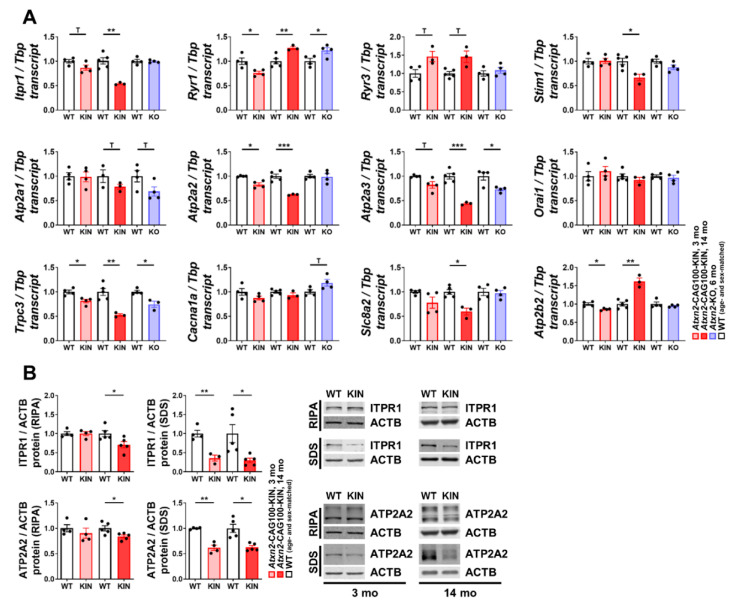
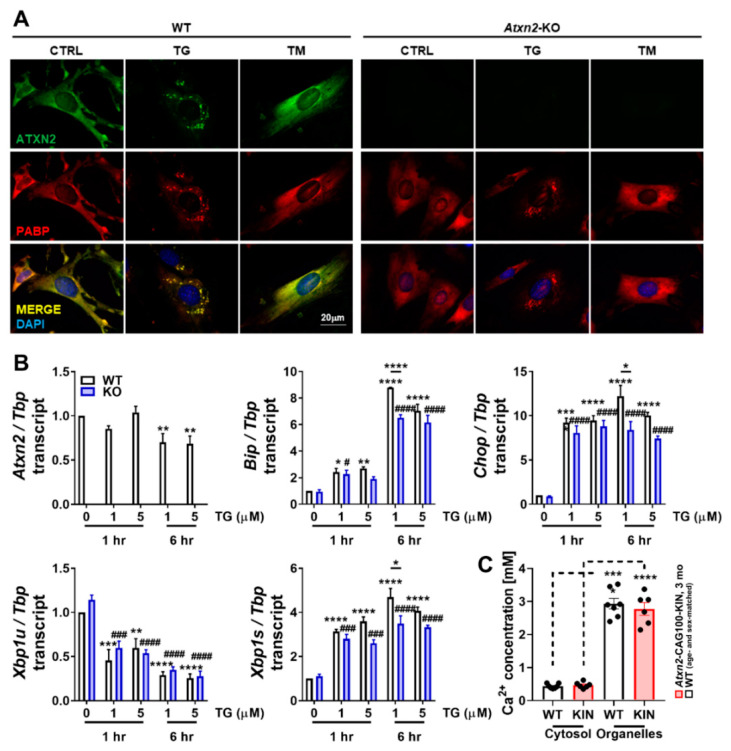
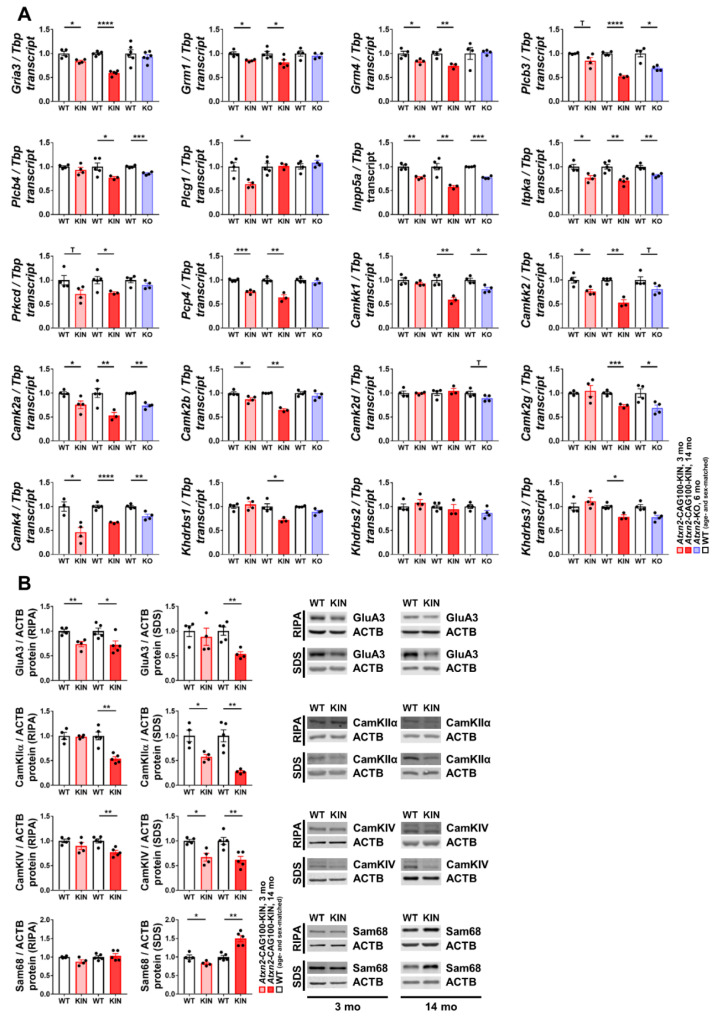
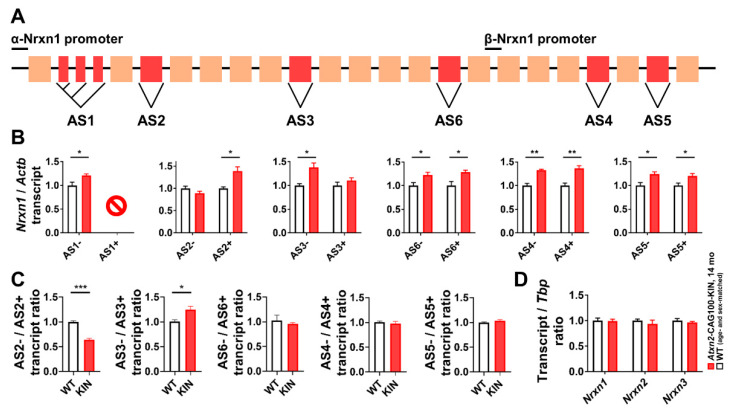
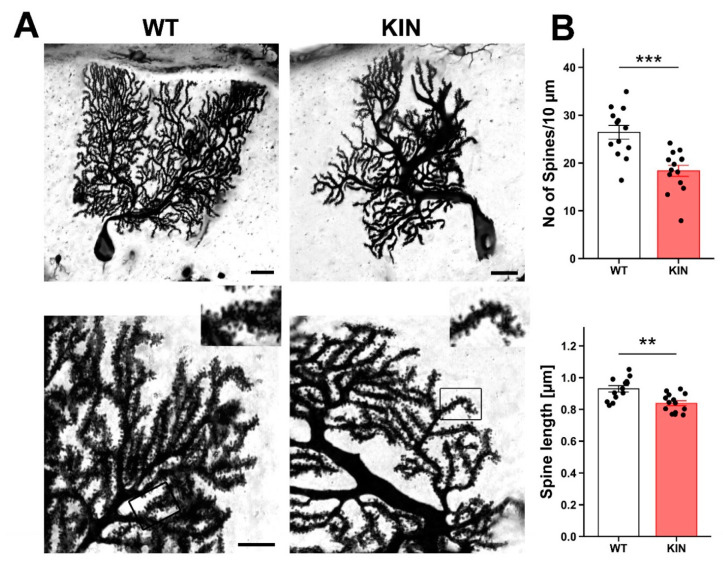
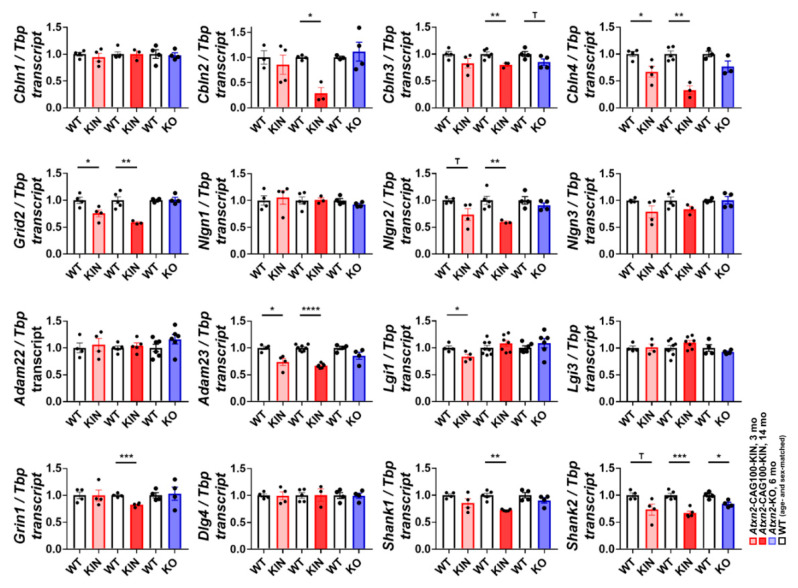
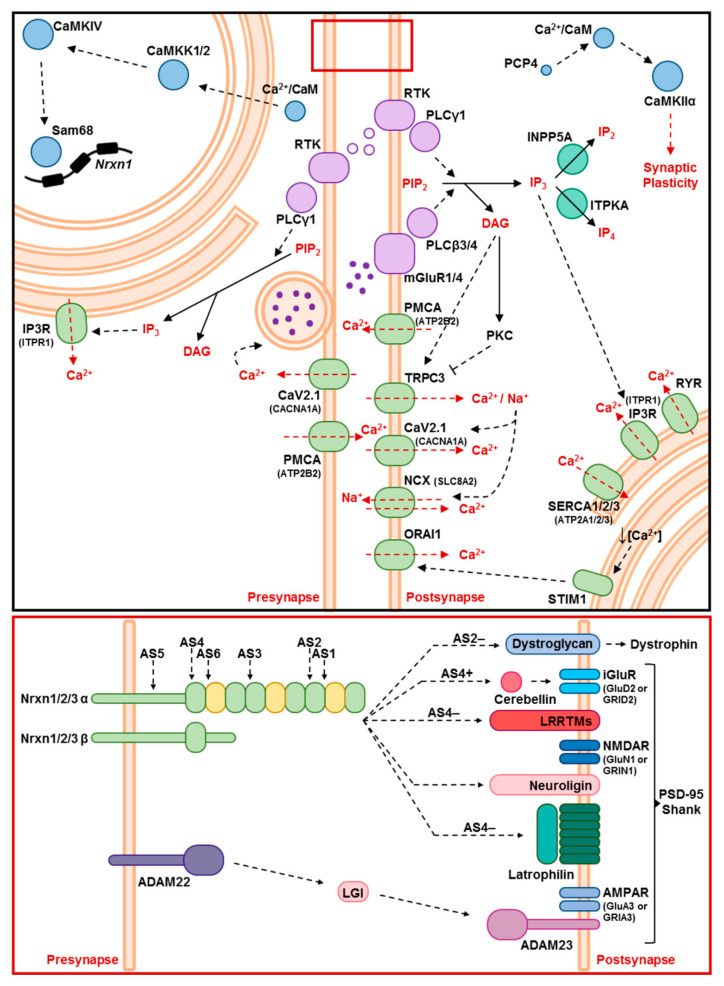
Spinocerebellar ataxia type 2 (SCA2) is caused by polyglutamine expansion in Ataxin-2 (ATXN2). This factor binds RNA/proteins to modify metabolism after stress, and to control calcium (Ca2+) homeostasis after stimuli. Cerebellar ataxias and corticospinal motor neuron degeneration are determined by gain/loss in ATXN2 function, so we aimed to identify key molecules in this atrophic process, as potential disease progression markers. Our Atxn2-CAG100-Knock-In mouse faithfully models features observed in patients at pre-onset, early and terminal stages. Here, its cerebellar global RNA profiling revealed downregulation of signaling cascades to precede motor deficits. Validation work at mRNA/protein level defined alterations that were independent of constant physiological ATXN2 functions, but specific for RNA/aggregation toxicity, and progressive across the short lifespan. The earliest changes were detected at three months among Ca2+ channels/transporters (Itpr1, Ryr3, Atp2a2, Atp2a3, Trpc3), IP3 metabolism (Plcg1, Inpp5a, Itpka), and Ca2+-Calmodulin dependent kinases (Camk2a, Camk4). CaMKIV-Sam68 control over alternative splicing of Nrxn1, an adhesion component of glutamatergic synapses between granule and Purkinje neurons, was found to be affected. Systematic screening of pre/post-synapse components, with dendrite morphology assessment, suggested early impairment of CamKIIα abundance together with the weakening of parallel fiber connectivity. These data reveal molecular changes due to ATXN2 pathology, primarily impacting excitability and communication.
Keywords: K-homology RNA-binding domain; amyotrophic lateral sclerosis (ALS); fragile-X-associated tremor-ataxia syndrome; fronto-temporal-lobar-dementia; inositol signaling; long-term potentiation; neurexin; spatial learning; synaptic plasticity; tauopathies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Pulst S.M., Nechiporuk A., Nechiporuk T., Gispert S., Chen X.N., Lopes-Cendes I., Pearlman S., Starkman S., Orozco-Diaz G., Lunkes A., et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 1996;14:269–276. doi: 10.1038/ng1196-269. - DOI - PubMed
-
- Sanpei K., Takano H., Igarashi S., Sato T., Oyake M., Sasaki H., Wakisaka A., Tashiro K., Ishida Y., Ikeuchi T., et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat. Genet. 1996;14:277–284. doi: 10.1038/ng1196-277. - DOI - PubMed
-
- Imbert G., Saudou F., Yvert G., Devys D., Trottier Y., Garnier J.M., Weber C., Mandel J.L., Cancel G., Abbas N., et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 1996;14:285–291. doi: 10.1038/ng1196-285. - DOI - PubMed
-
- Velázquez-Pérez L., Seifried C., Santos-Falcón N., Abele M., Ziemann U., Almaguer L.E., Martínez-Góngora E., Sánchez-Cruz G., Canales N., Pérez-González R., et al. Saccade velocity is controlled by polyglutamine size in spinocerebellar ataxia 2. Ann. Neurol. 2004;56:444–447. doi: 10.1002/ana.20220. - DOI - PubMed
-
- Velázquez-Pérez L., Rodríguez-Labrada R., Canales-Ochoa N., Montero J.M., Sánchez-Cruz G., Aguilera-Rodríguez R., Almaguer-Mederos L.E., Laffita-Mesa J.M. Progression of early features of spinocerebellar ataxia type 2 in individuals at risk: A longitudinal study. Lancet Neurol. 2014;13:482–489. doi: 10.1016/S1474-4422(14)70027-4. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
