

Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence
- PMID: 32932888
- PMCID: PMC7563273
- DOI: 10.3390/jcm9092934
Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence
Abstract
Acute myeloid leukemia (AML) is a highly heterogeneous hematopoietic malignancy characterized by excessive proliferation and accumulation of immature myeloid blasts in the bone marrow. AML has a very poor 5-year survival rate of just 16% in the UK; hence, more efficacious, tolerable, and targeted therapy is required. Persistent leukemia stem cell (LSC) populations underlie patient relapse and development of resistance to therapy. Identification of critical oncogenic signaling pathways in AML LSC may provide new avenues for novel therapeutic strategies. The phosphatidylinositol-3-kinase (PI3K)/Akt and the mammalian target of rapamycin (mTOR) signaling pathway, is often hyperactivated in AML, required to sustain the oncogenic potential of LSCs. Growing evidence suggests that targeting key components of this pathway may represent an effective treatment to kill AML LSCs. Despite this, accruing significant body of scientific knowledge, PI3K/Akt/mTOR inhibitors have not translated into clinical practice. In this article, we review the laboratory-based evidence of the critical role of PI3K/Akt/mTOR pathway in AML, and outcomes from current clinical studies using PI3K/Akt/mTOR inhibitors. Based on these results, we discuss the putative mechanisms of resistance to PI3K/Akt/mTOR inhibition, offering rationale for potential candidate combination therapies incorporating PI3K/Akt/mTOR inhibitors for precision medicine in AML.
Keywords: AML; LSC; PI3K/Akt; combination treatment strategy; drug resistance; mTOR; targeted therapy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

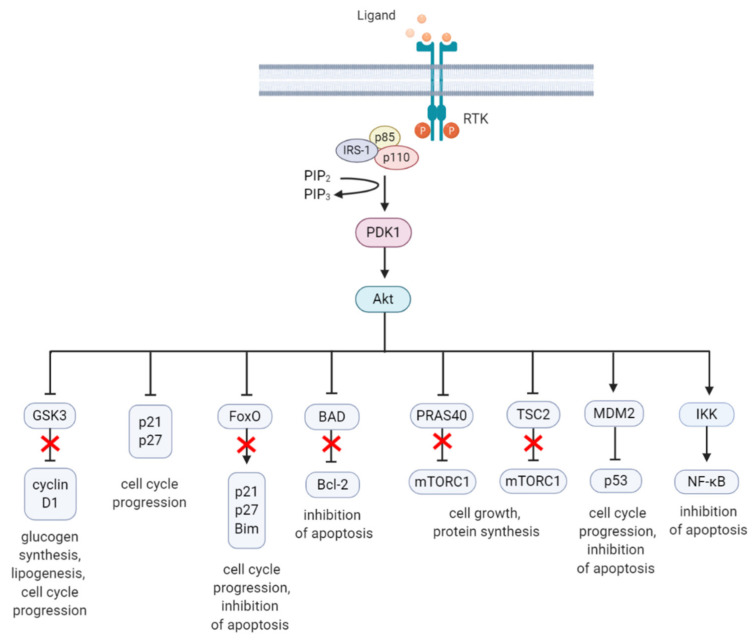
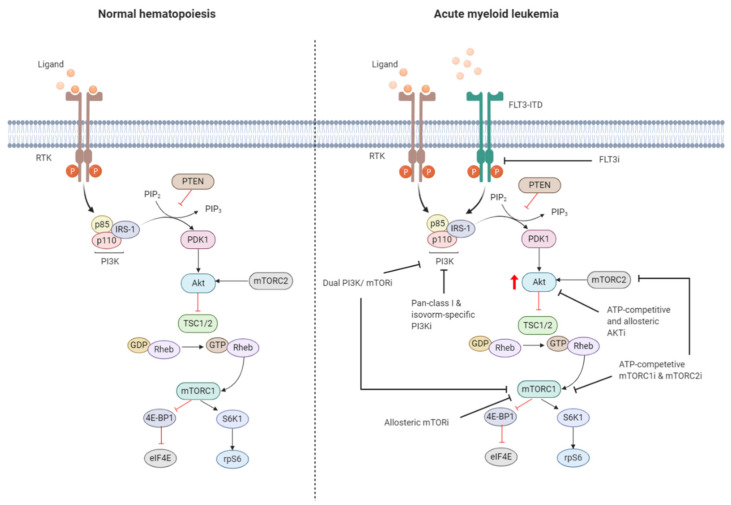
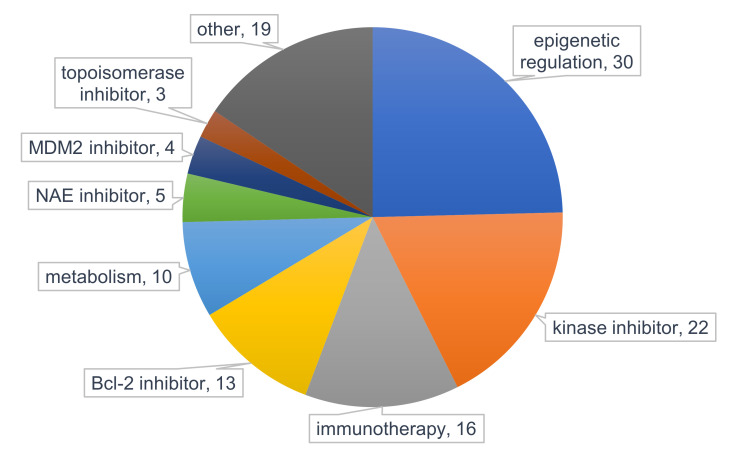
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
