

This is a preprint.
Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition
- PMID: 32935107
- PMCID: PMC7491521
- DOI: 10.1101/2020.09.10.292078
Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition
Update in
-
Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition.Cell Host Microbe. 2021 Jan 13;29(1):44-57.e9. doi: 10.1016/j.chom.2020.11.007. Epub 2020 Nov 19. Cell Host Microbe. 2021. PMID: 33259788 Free PMC article.
Abstract
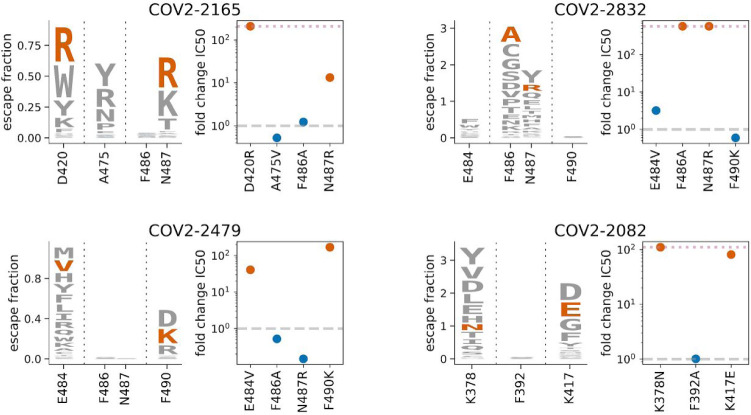
Antibodies targeting the SARS-CoV-2 spike receptor-binding domain (RBD) are being developed as therapeutics and make a major contribution to the neutralizing antibody response elicited by infection. Here, we describe a deep mutational scanning method to map how all amino-acid mutations in the RBD affect antibody binding, and apply this method to 10 human monoclonal antibodies. The escape mutations cluster on several surfaces of the RBD that broadly correspond to structurally defined antibody epitopes. However, even antibodies targeting the same RBD surface often have distinct escape mutations. The complete escape maps predict which mutations are selected during viral growth in the presence of single antibodies, and enable us to design escape-resistant antibody cocktails-including cocktails of antibodies that compete for binding to the same surface of the RBD but have different escape mutations. Therefore, complete escape-mutation maps enable rational design of antibody therapeutics and assessment of the antigenic consequences of viral evolution.
Conflict of interest statement
Declarations of Interests J.E.C. has served as a consultant for Sanofi; is on the Scientific Advisory Boards of CompuVax and Meissa Vaccines; is a recipient of previous unrelated research grants from Moderna and Sanofi; and is a founder of IDBiologics. Vanderbilt University has applied for patents concerning SARS-CoV-2 antibodies analyzed in this work. S.P.J.W. and P.W.R. have filed a disclosure with Washington University for the recombinant VSV. The other authors declare no competing interests.
Figures





References
-
- Amir E.-A.D., Davis K.L., Tadmor M.D., Simonds E.F., Levine J.H., Bendall S.C., Shenfeld D.K., Krishnaswamy S., Nolan G.P., and Pe’er D. (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 31, 545–552. - PMC - PubMed
-
- Barnes C.O., West A.P. Jr, Huey-Tubman K.E., Hoffmann M.A.G., Sharaf N.G., Hoffman P.R., Koranda N., Gristick H.B., Gaebler C., Muecksch F., et al. (2020a). Structures of human antibodies bound to SARS-CoV-2 Spike reveal common epitopes and recurrent features of antibodies. Cell 182, 828–842.e16. - PMC - PubMed
-
- Barnes C.O., Jette C.A., Abernathy M.E., Dam K.-M.A., Esswein S.R., Gristick H.B., Malyutin A.G., Sharaf N.G., Huey-Tubman K.E., Lee Y.E., et al. (2020b). Structural classification of neutralizing antibodies against the SARS-CoV-2 spike receptor-binding domain suggests vaccine and therapeutic strategies. bioRxiv 2020.08.30.273920.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous