

New developments in RiPP discovery, enzymology and engineering
- PMID: 32935693
- PMCID: PMC7864896
- DOI: 10.1039/d0np00027b
New developments in RiPP discovery, enzymology and engineering
Abstract
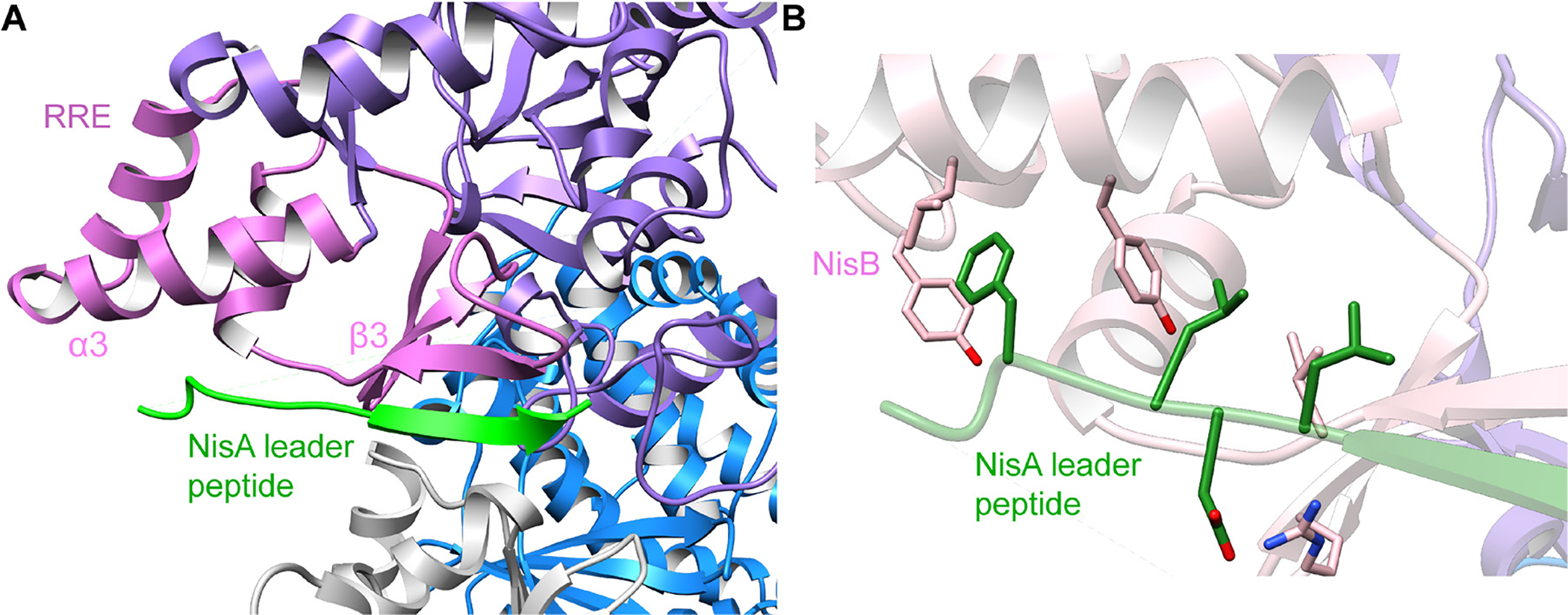
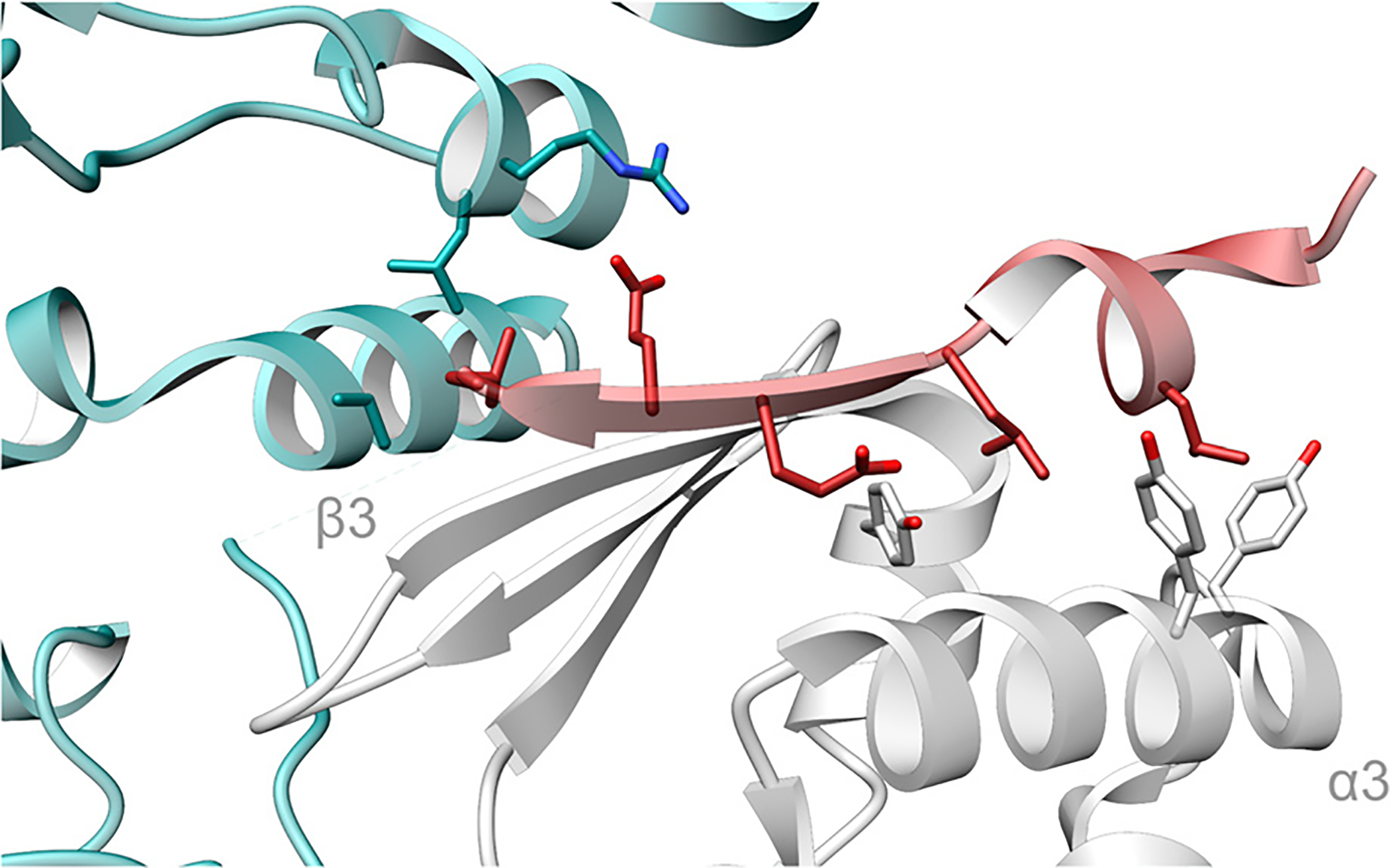
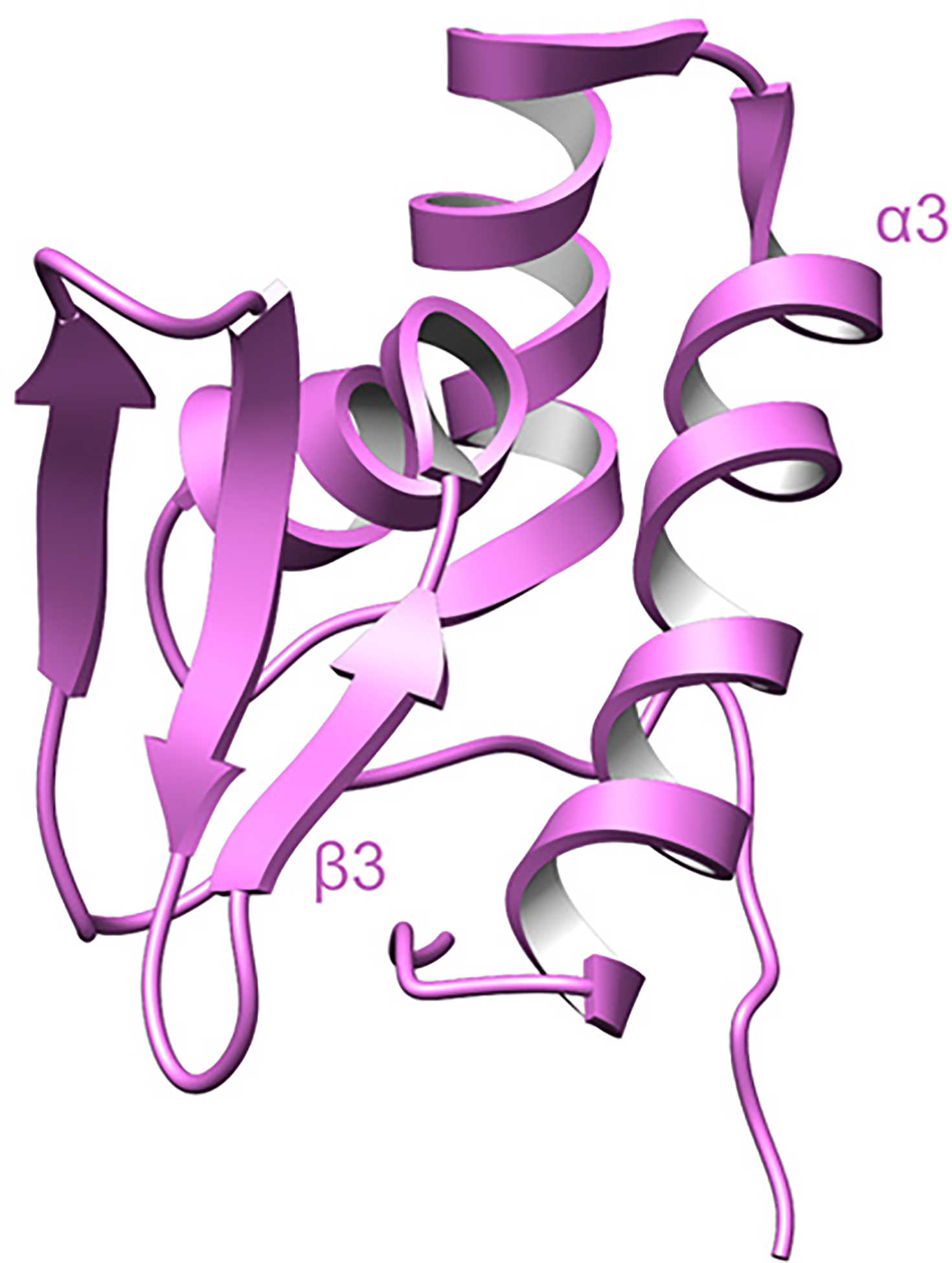
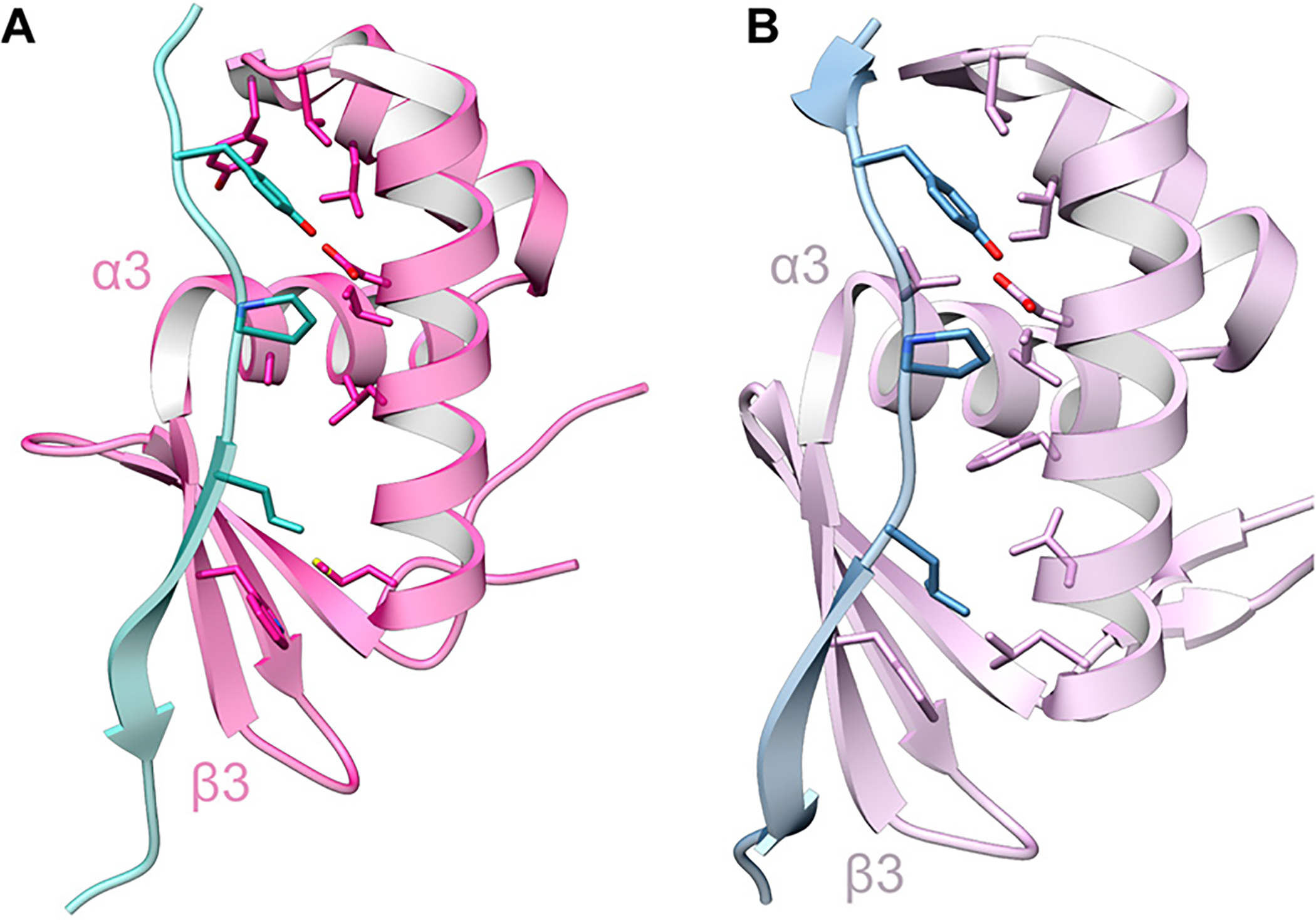
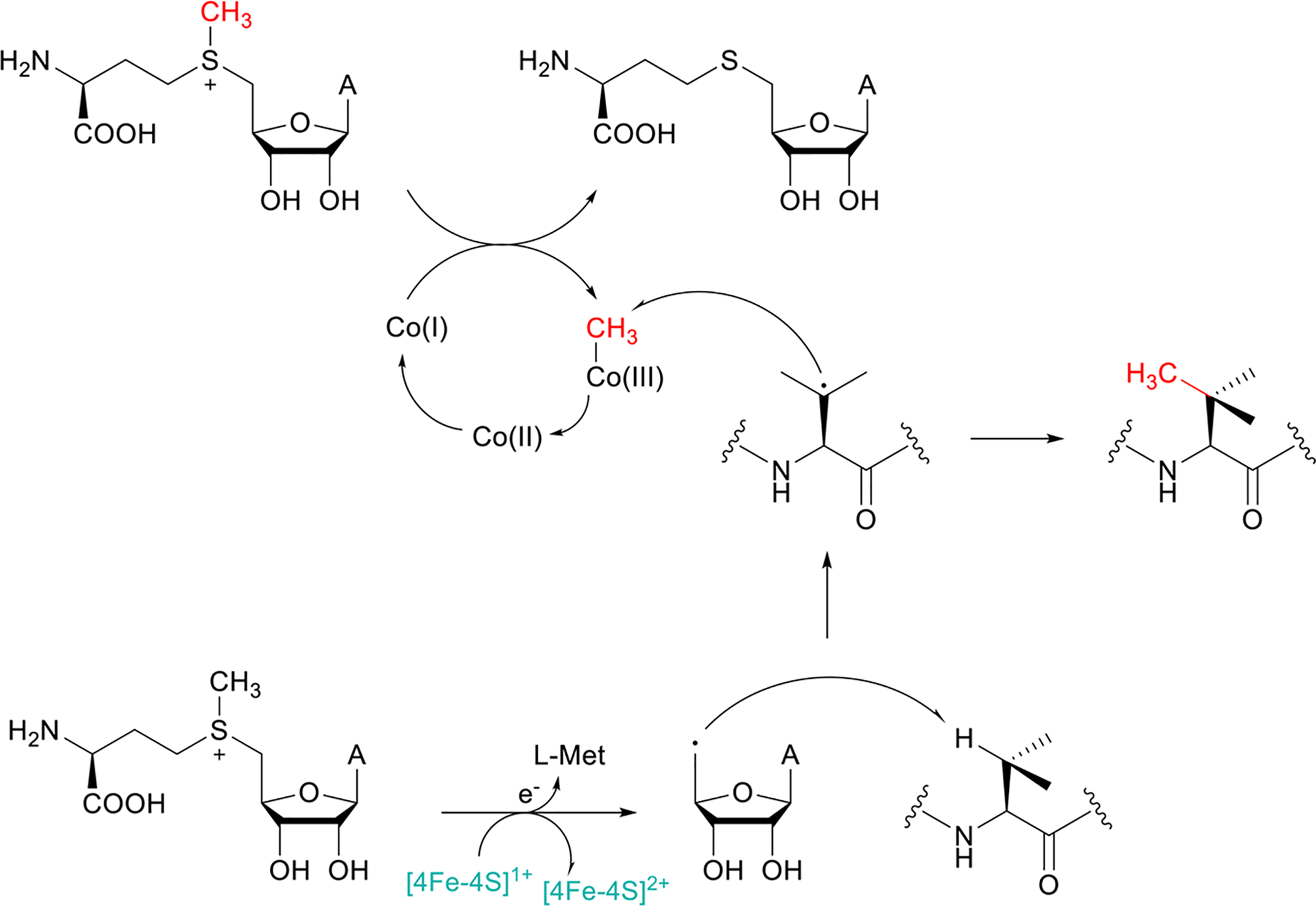
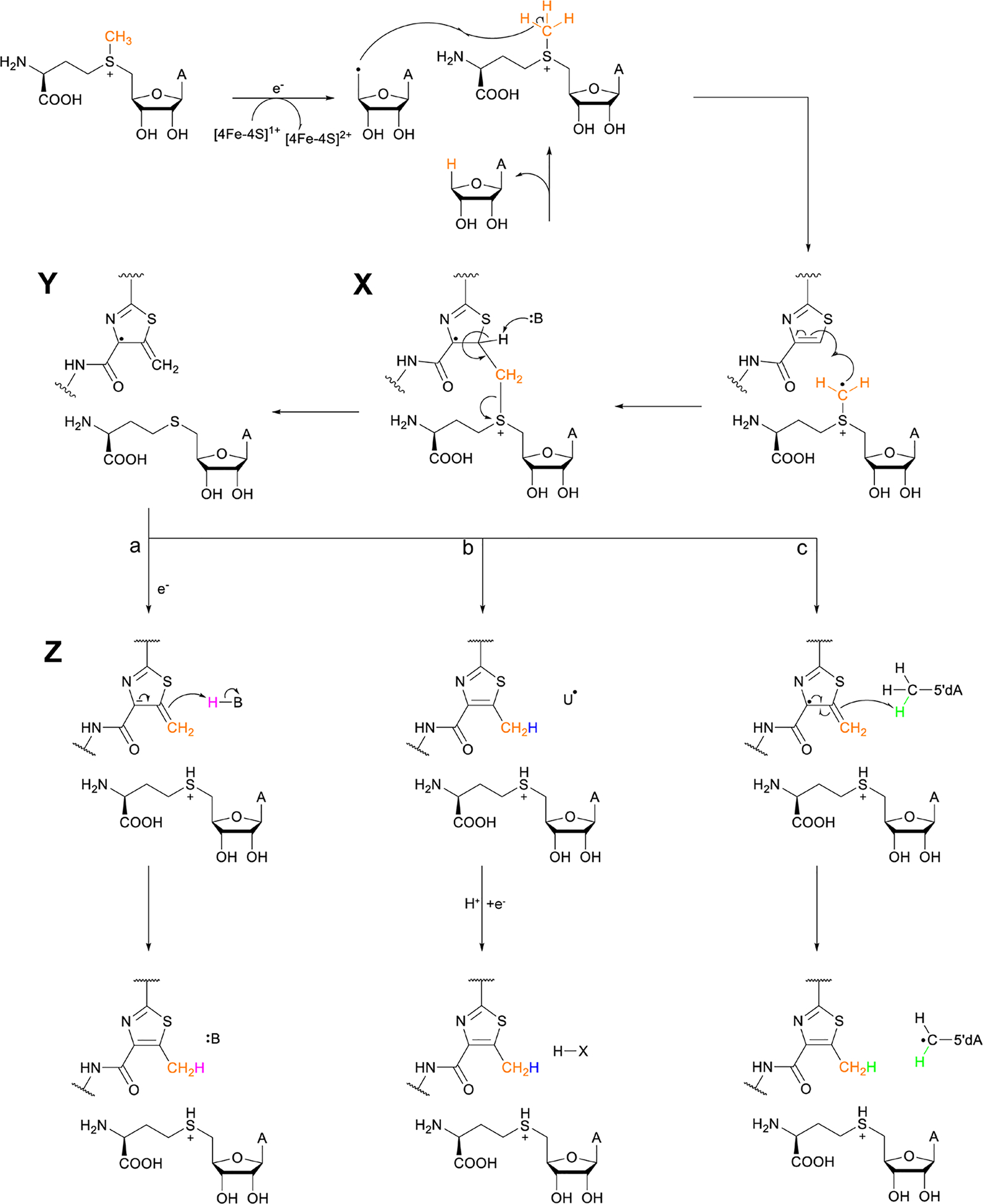
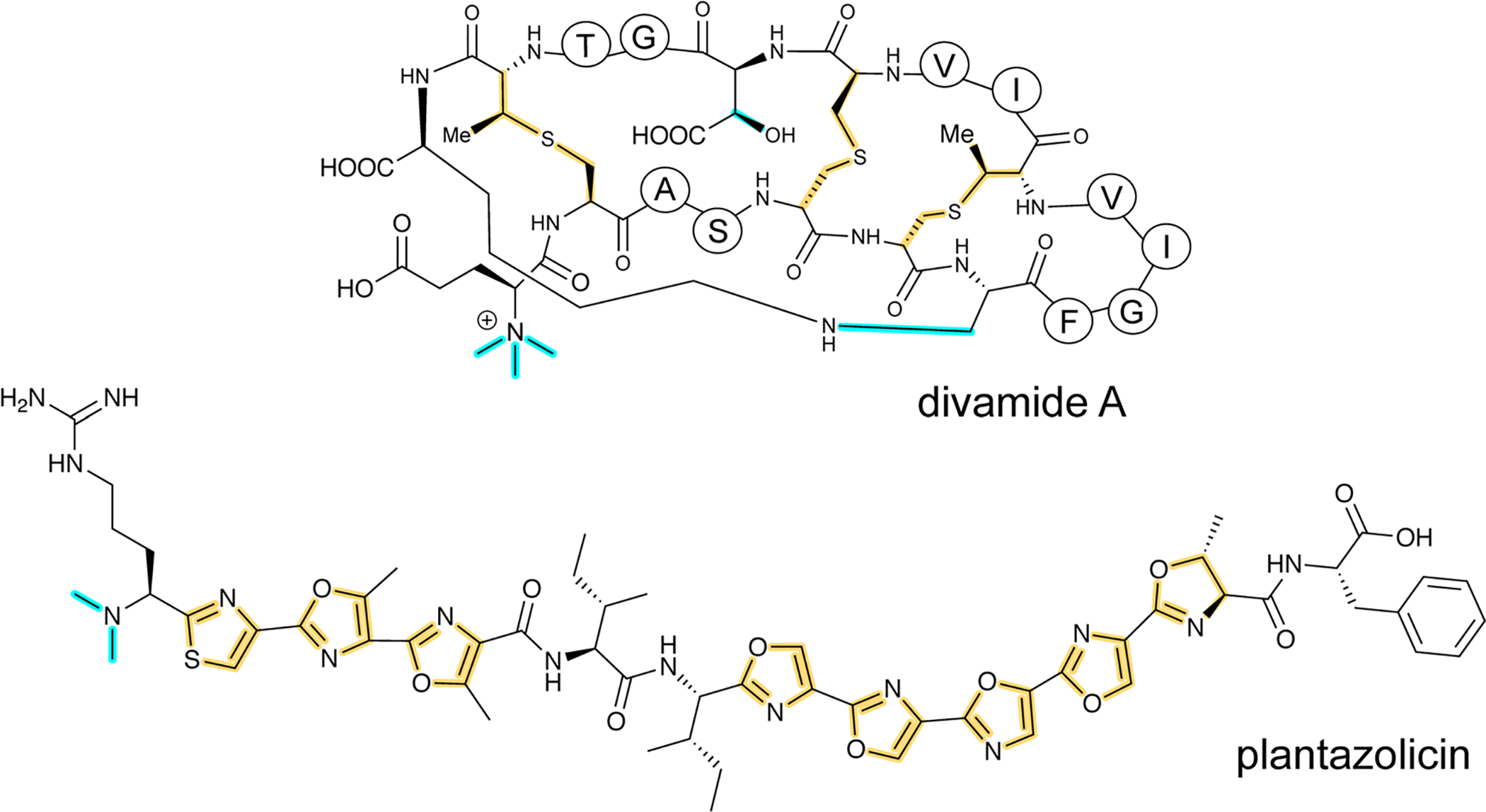
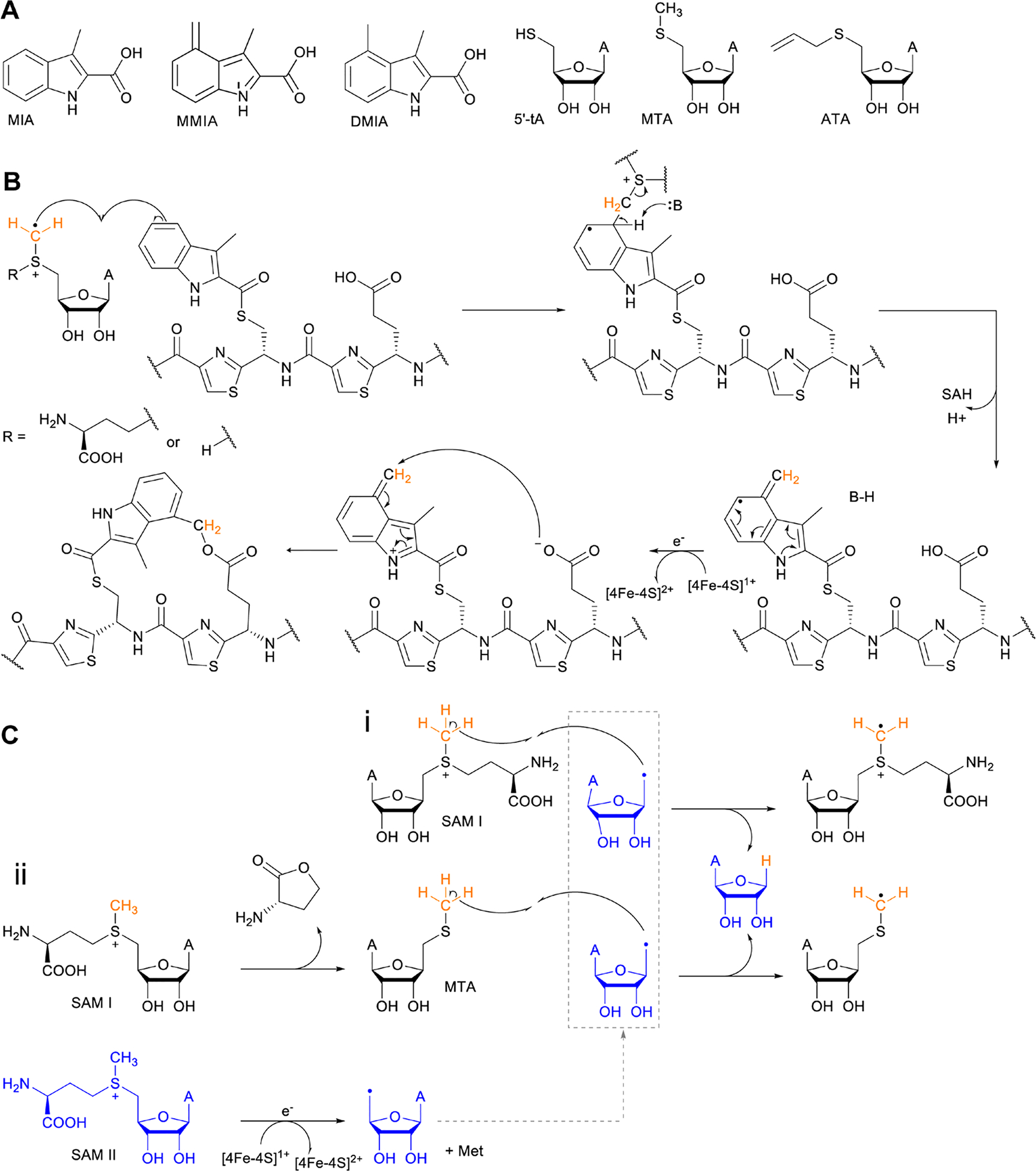
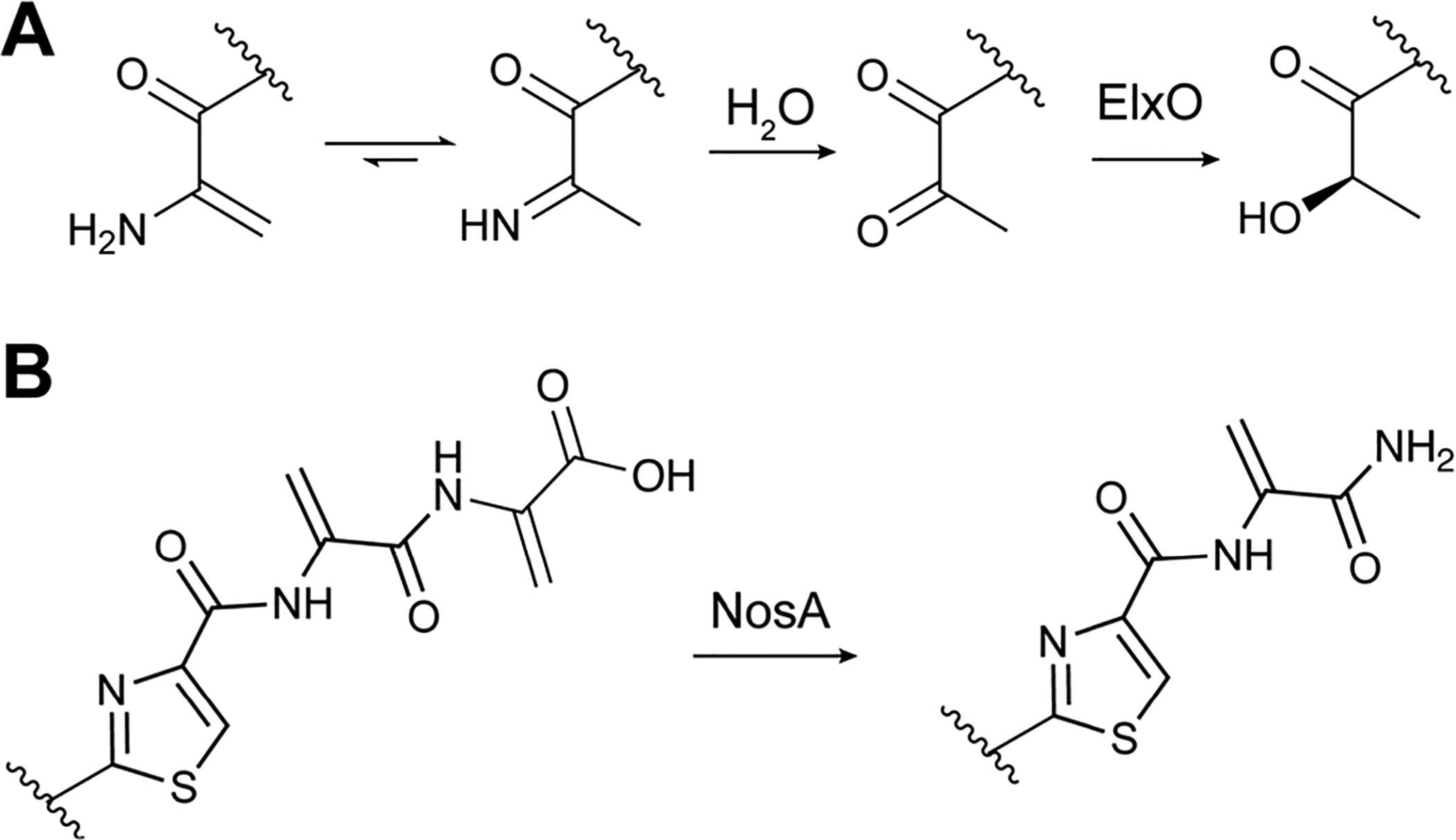
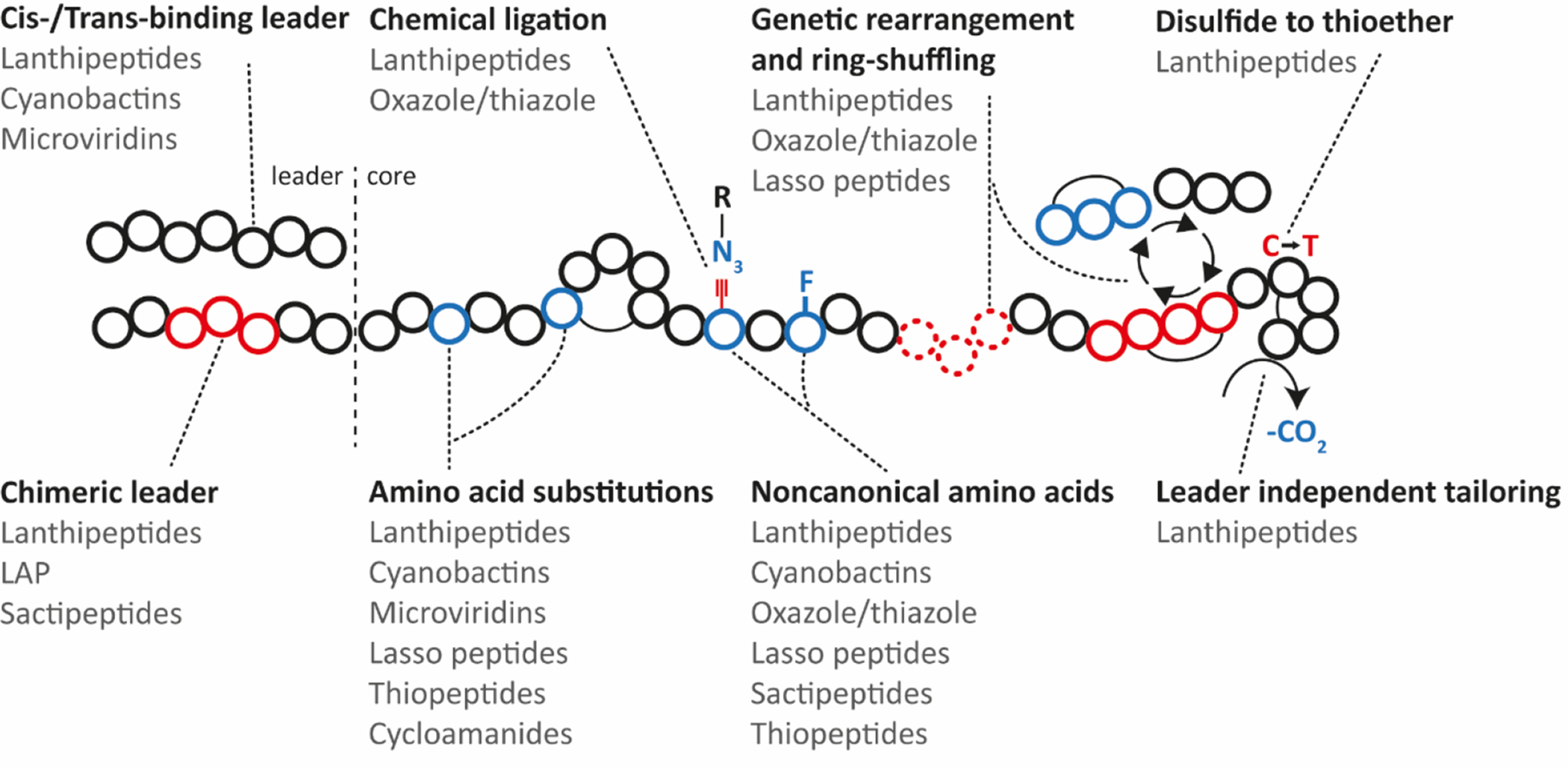
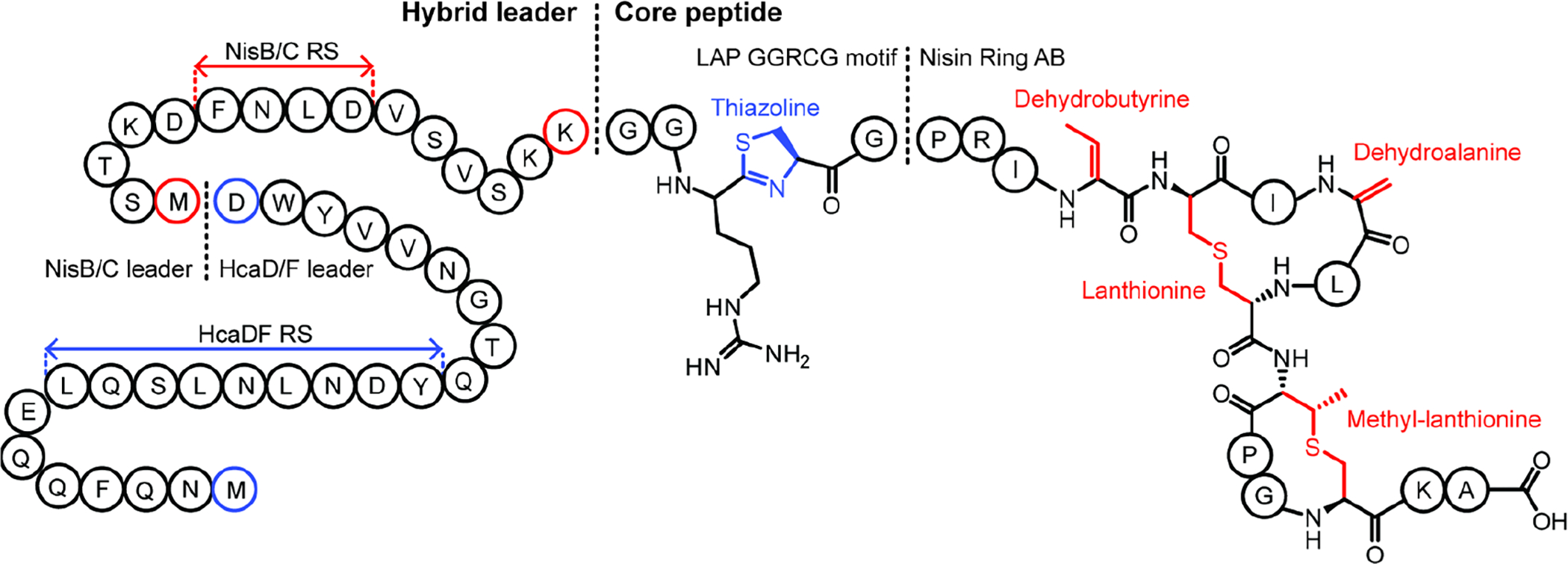
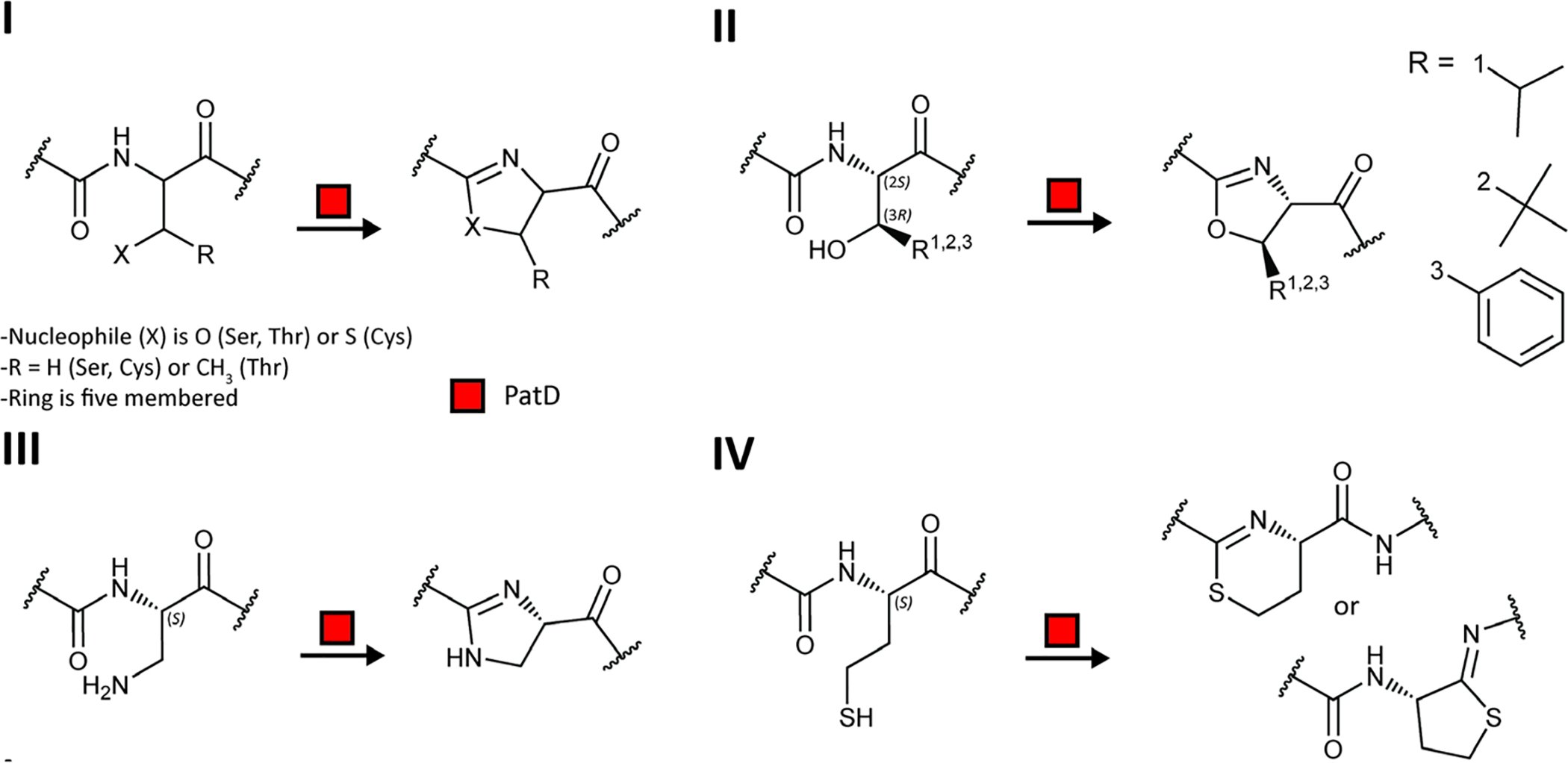
Covering: up to June 2020Ribosomally-synthesized and post-translationally modified peptides (RiPPs) are a large group of natural products. A community-driven review in 2013 described the emerging commonalities in the biosynthesis of RiPPs and the opportunities they offered for bioengineering and genome mining. Since then, the field has seen tremendous advances in understanding of the mechanisms by which nature assembles these compounds, in engineering their biosynthetic machinery for a wide range of applications, and in the discovery of entirely new RiPP families using bioinformatic tools developed specifically for this compound class. The First International Conference on RiPPs was held in 2019, and the meeting participants assembled the current review describing new developments since 2013. The review discusses the new classes of RiPPs that have been discovered, the advances in our understanding of the installation of both primary and secondary post-translational modifications, and the mechanisms by which the enzymes recognize the leader peptides in their substrates. In addition, genome mining tools used for RiPP discovery are discussed as well as various strategies for RiPP engineering. An outlook section presents directions for future research.
Figures
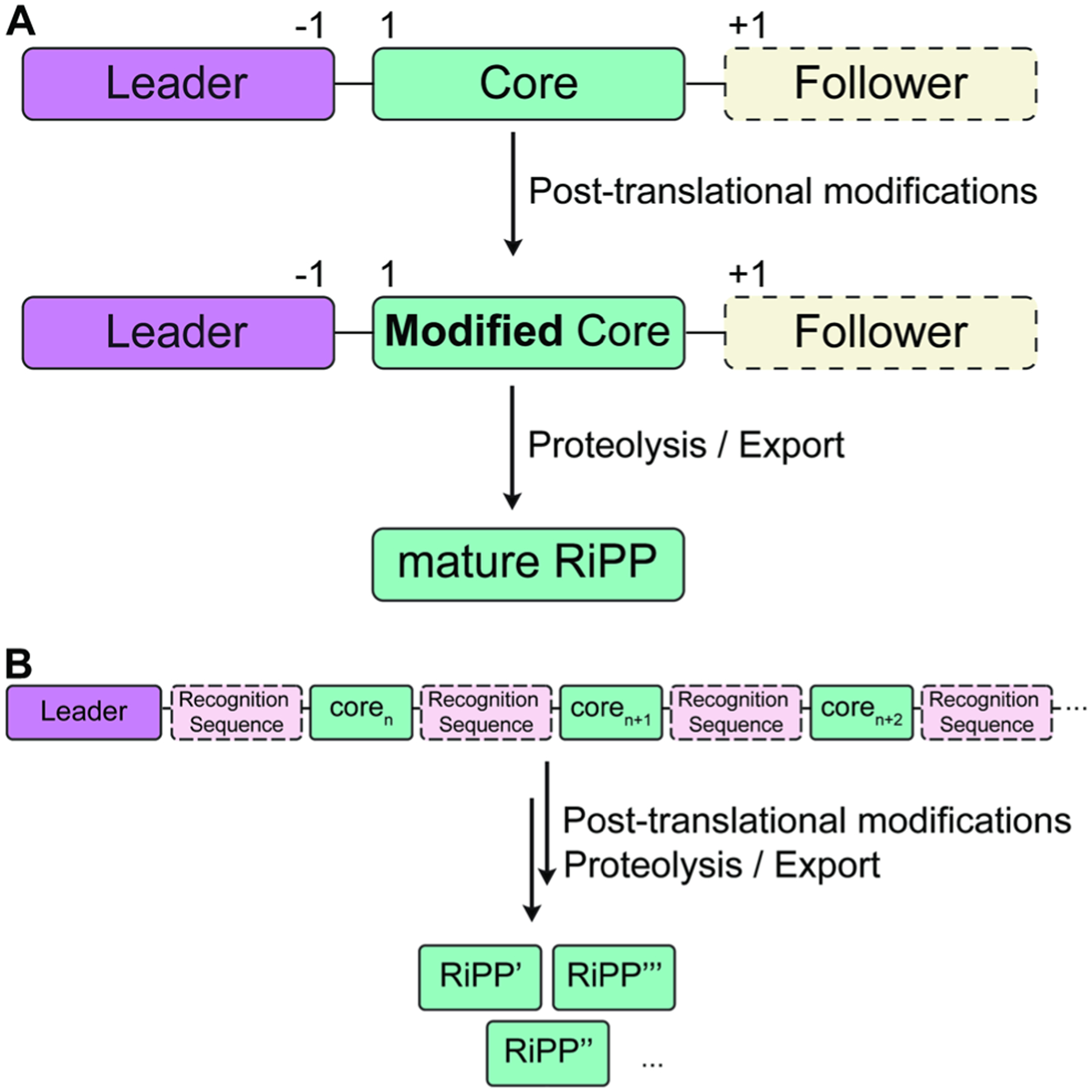
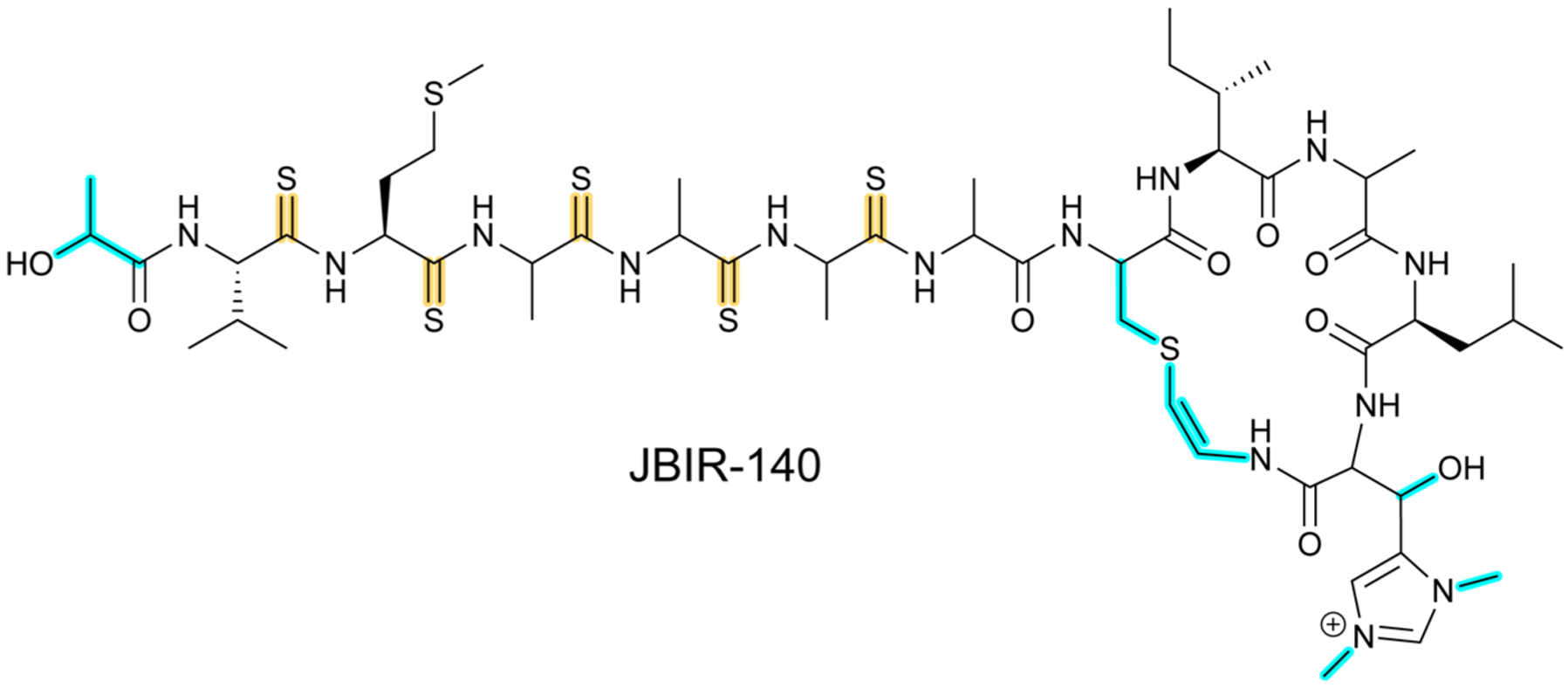
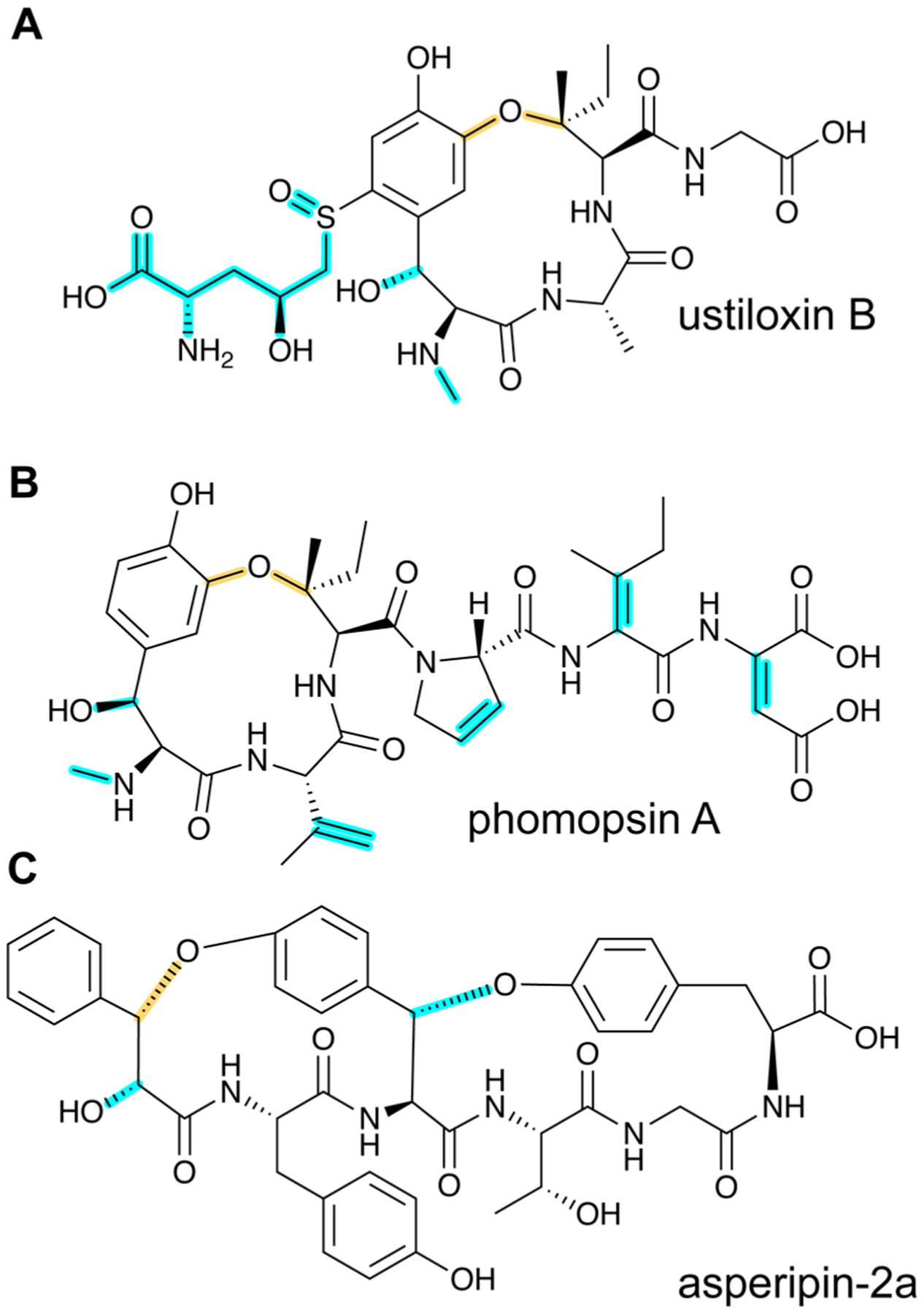
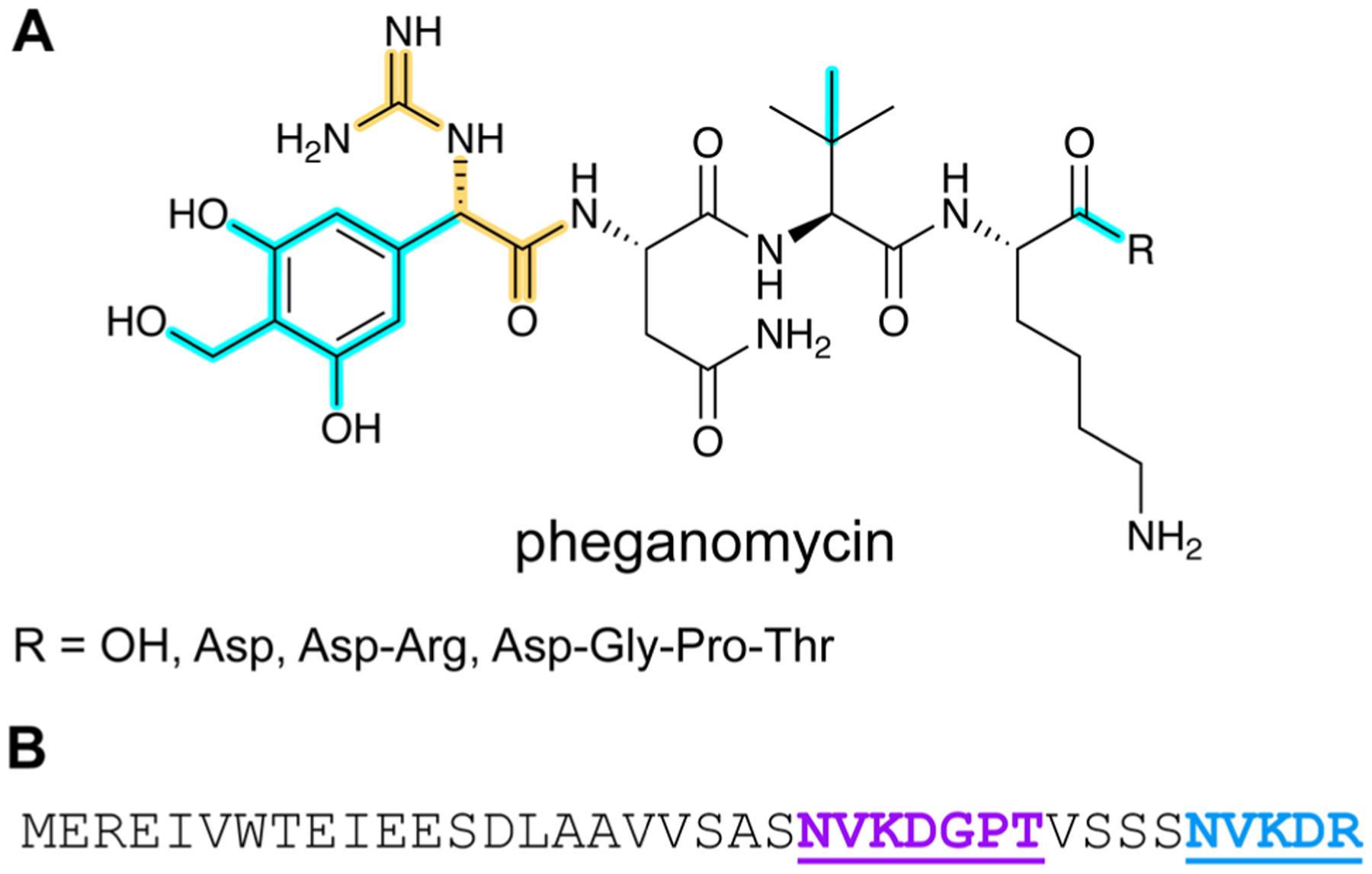
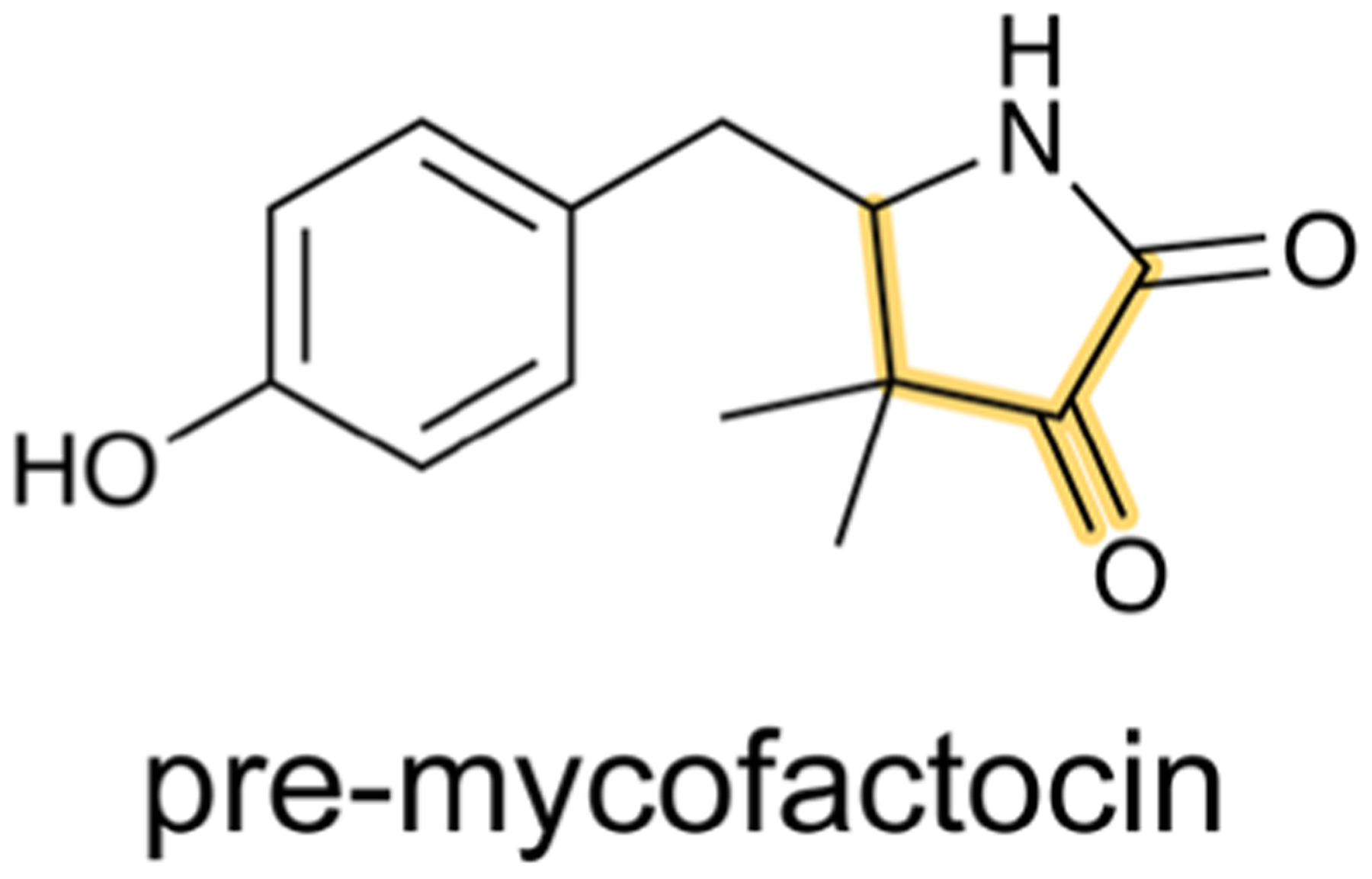
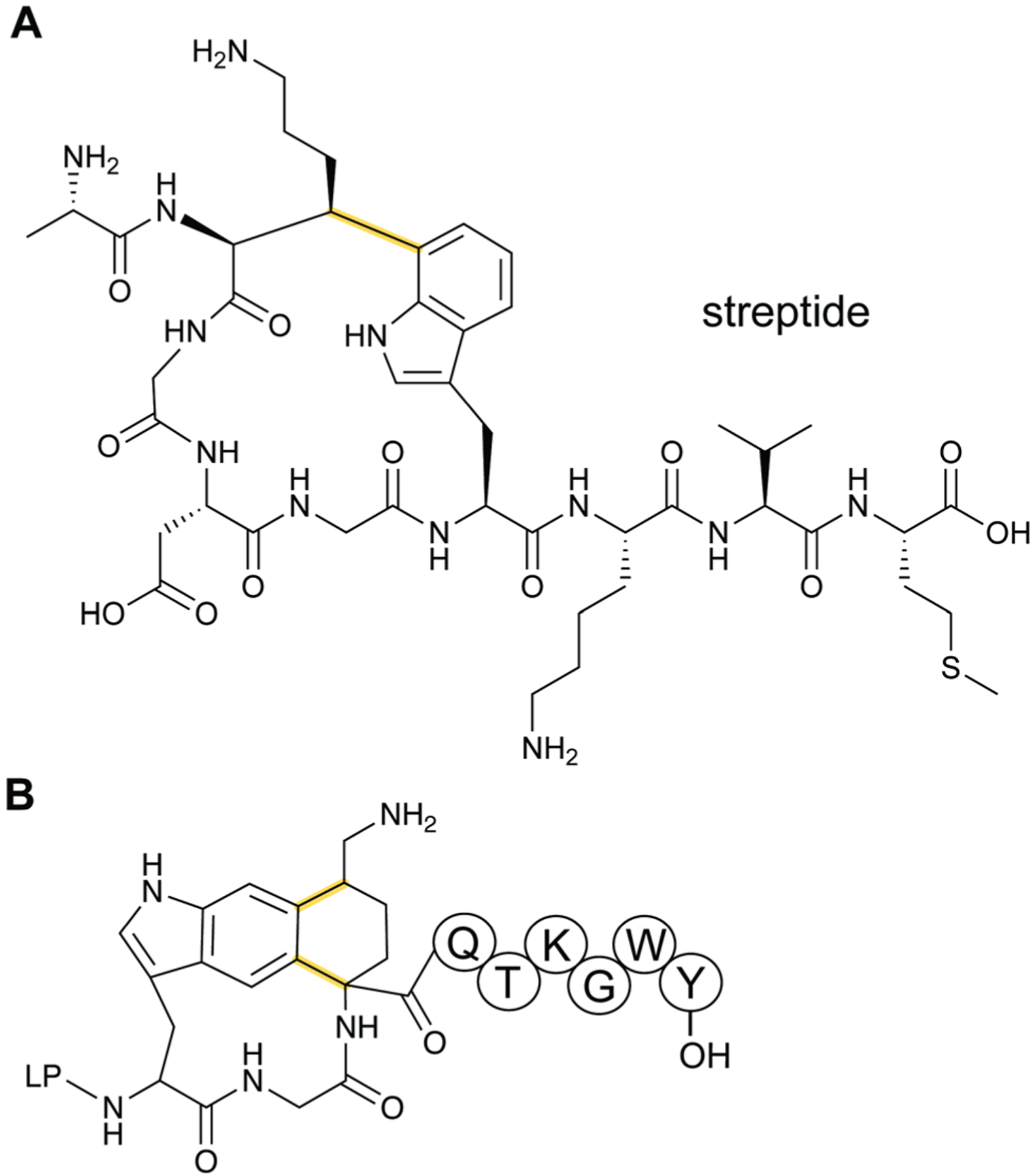
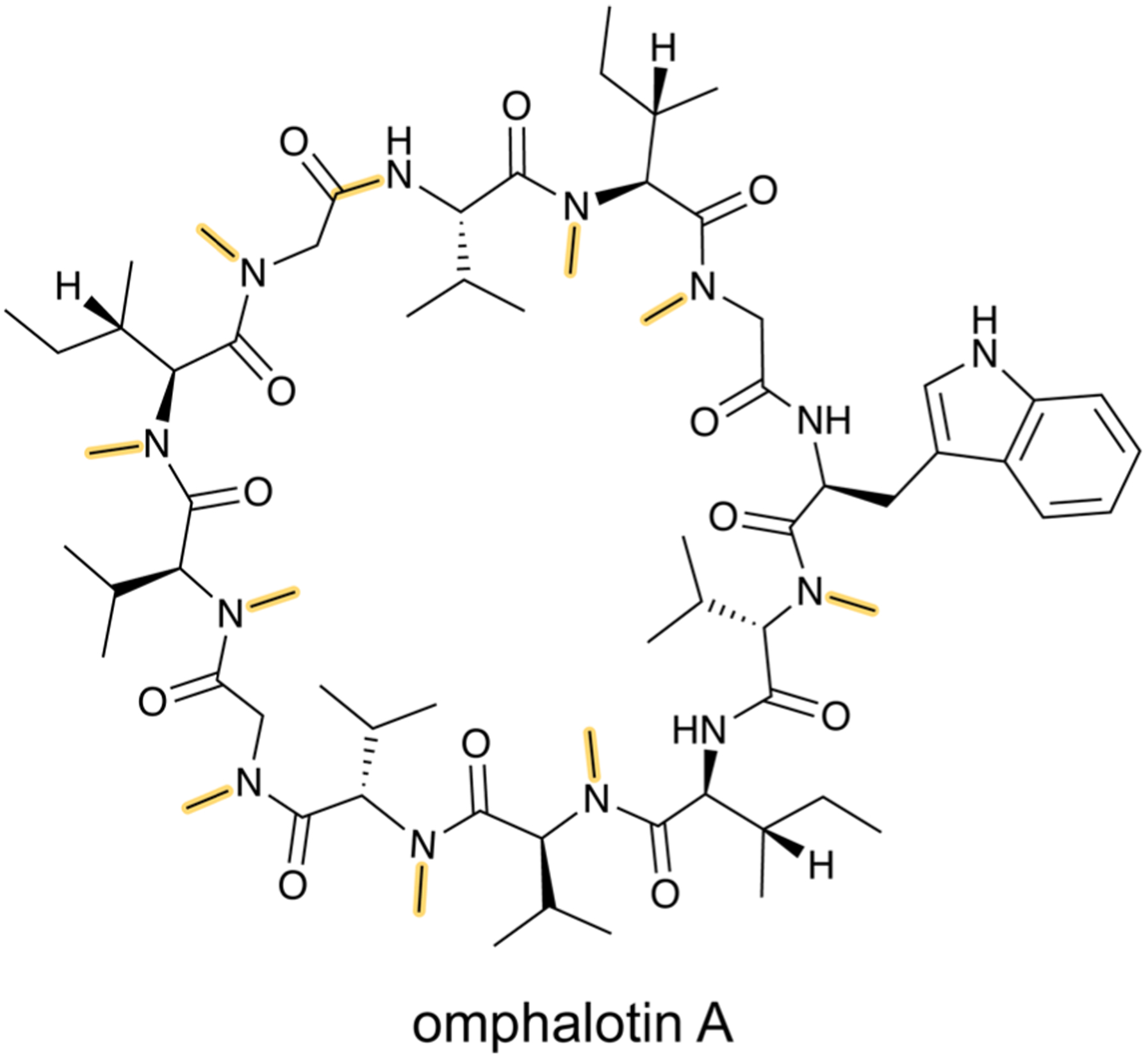
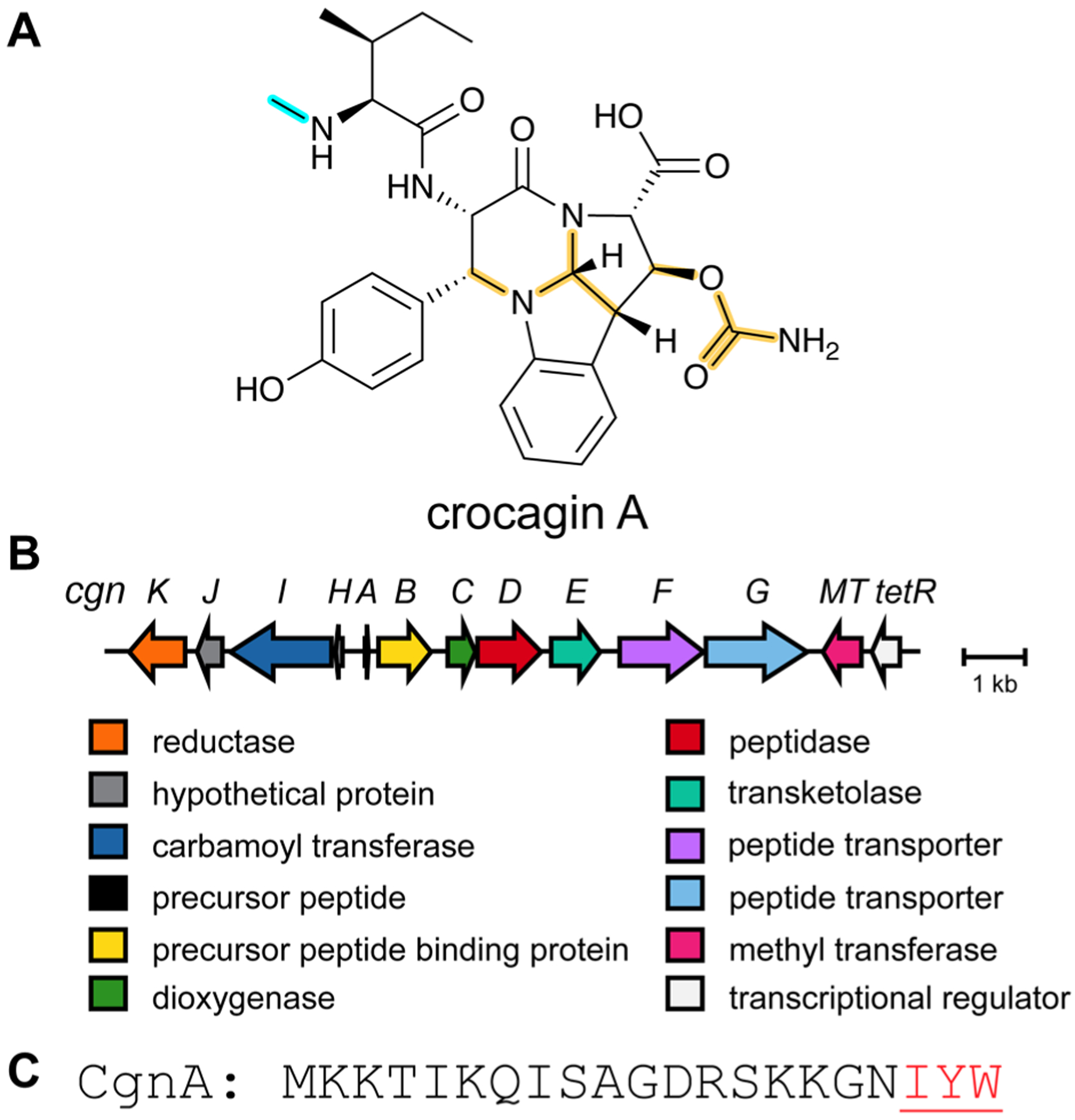
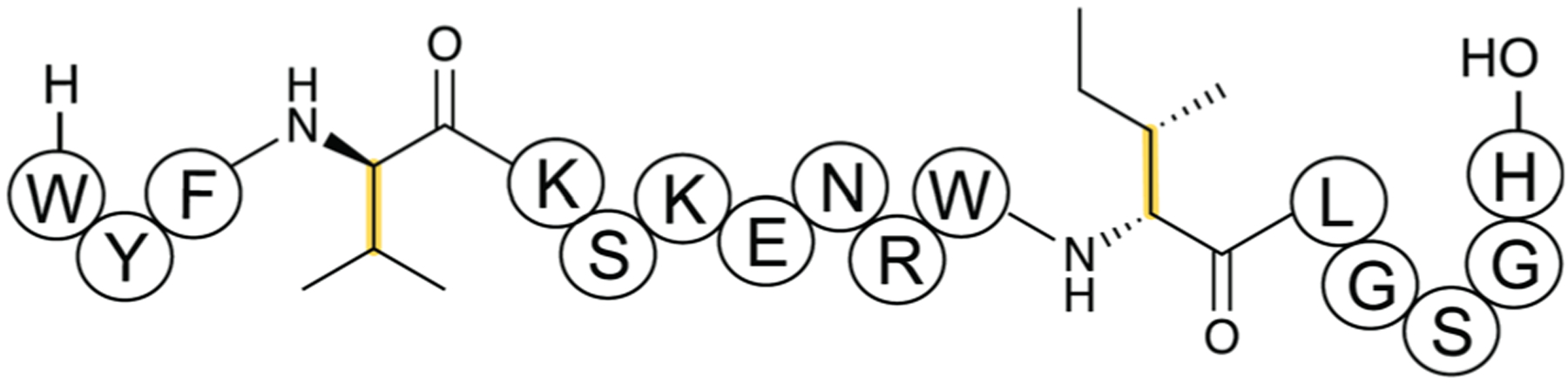
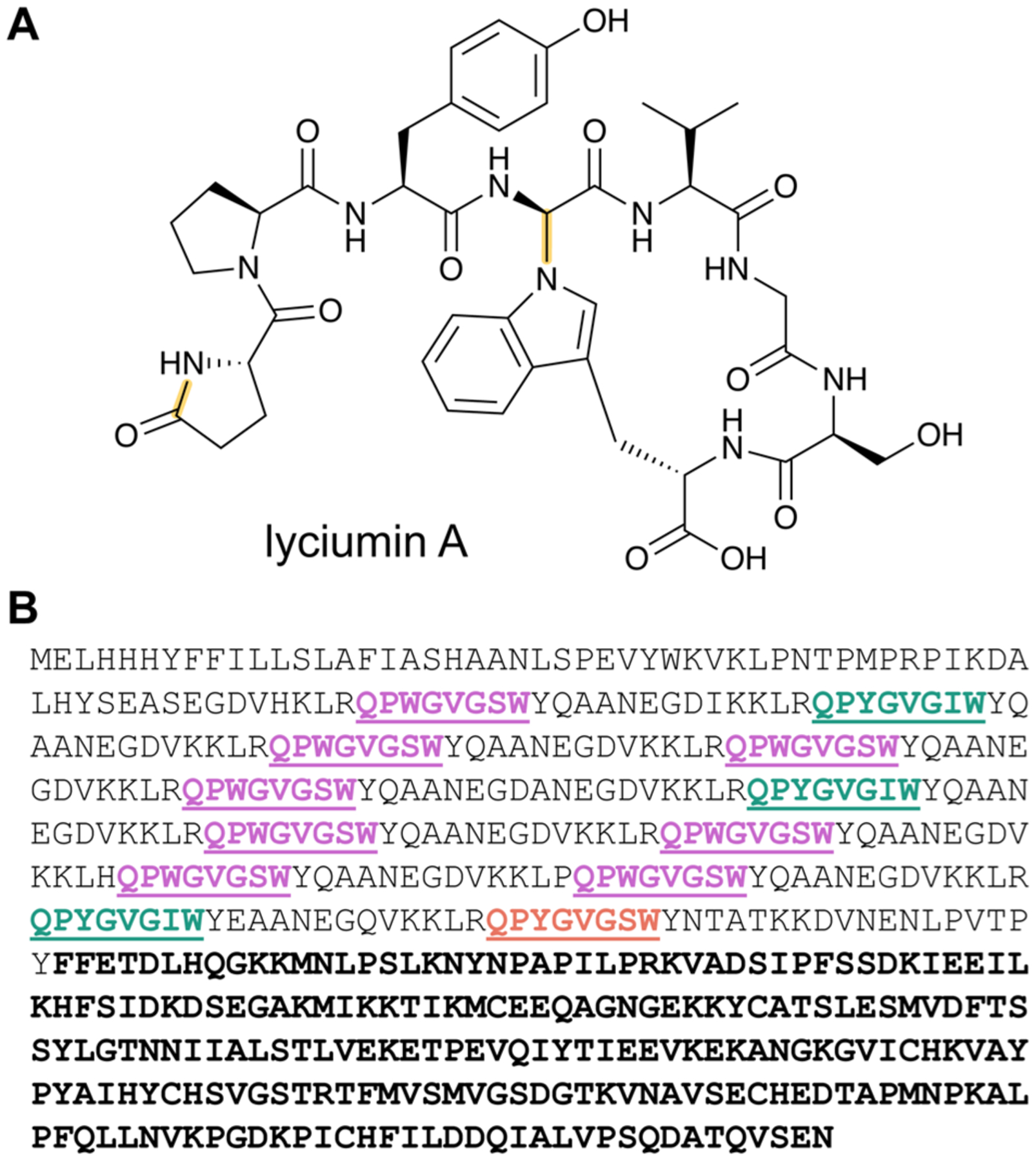
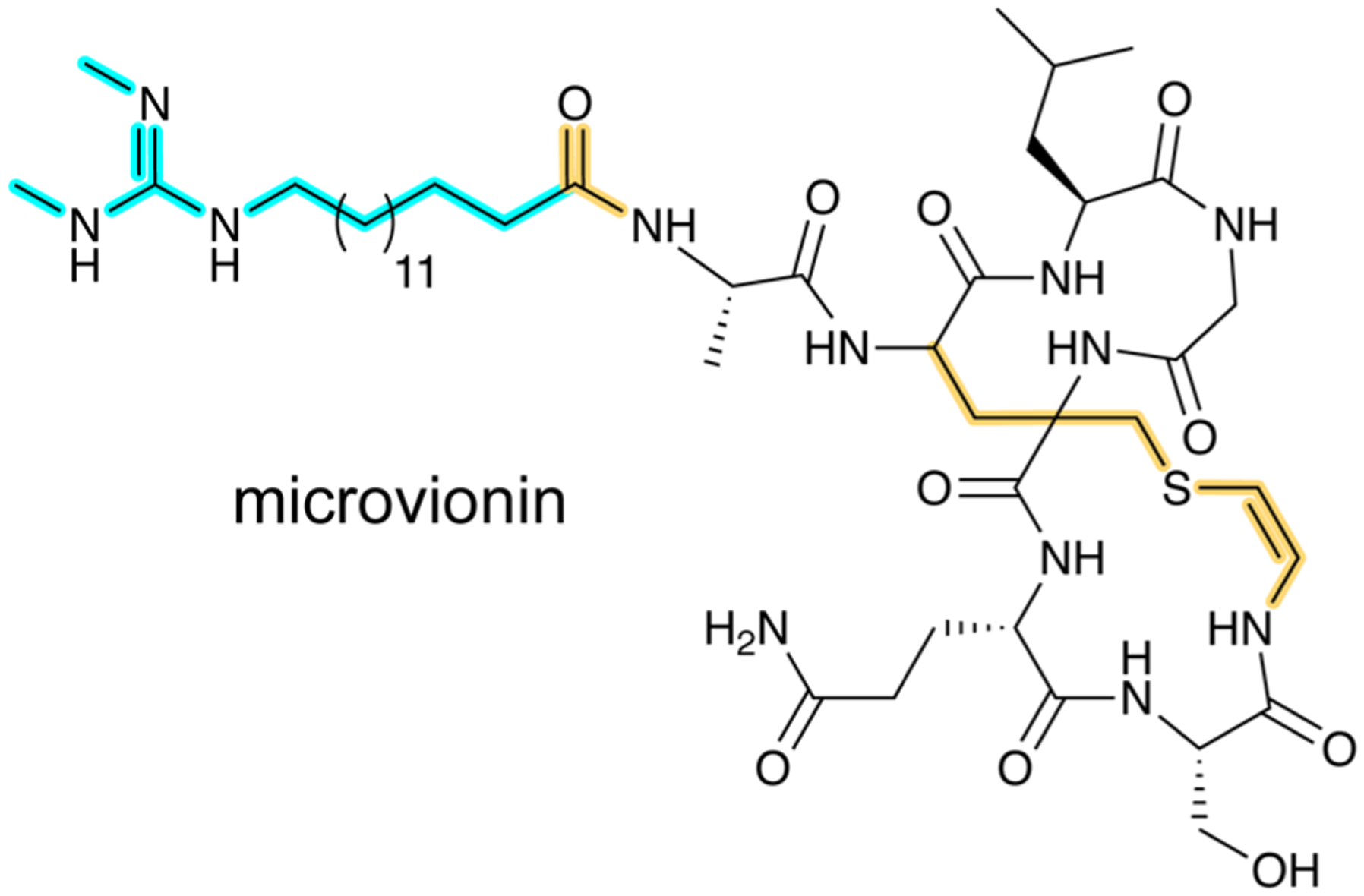
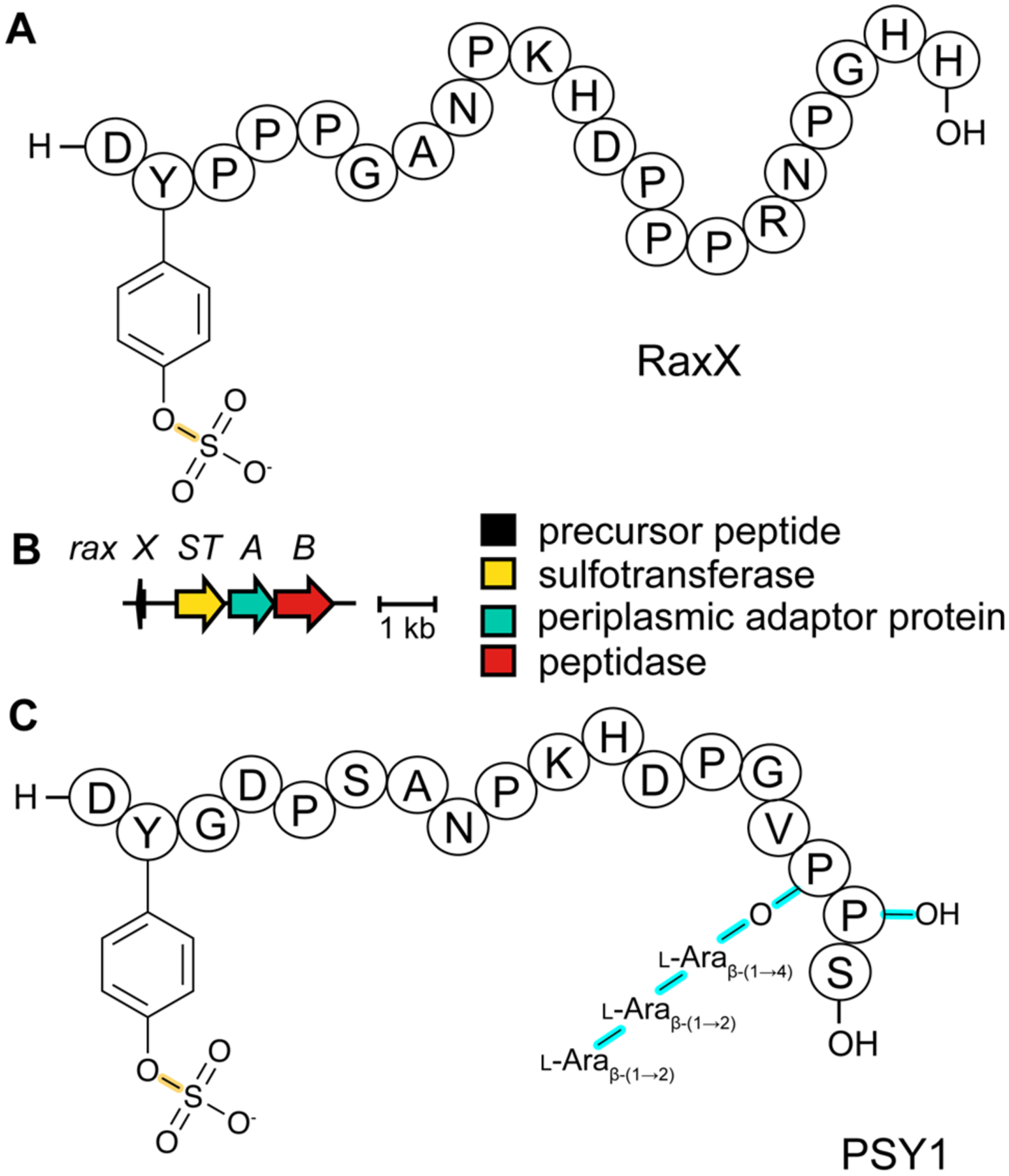
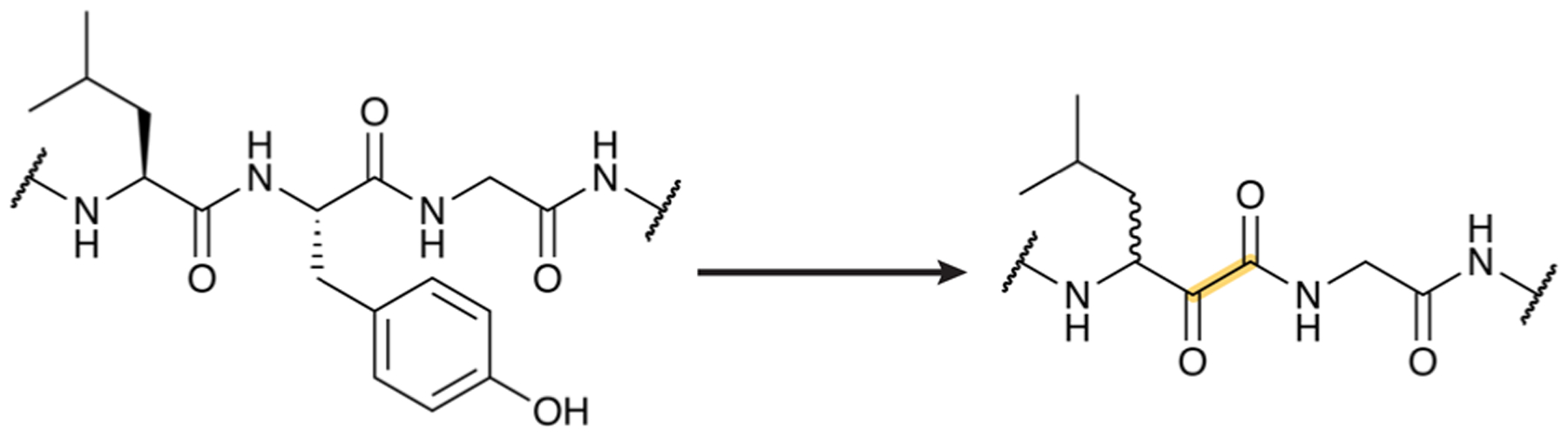
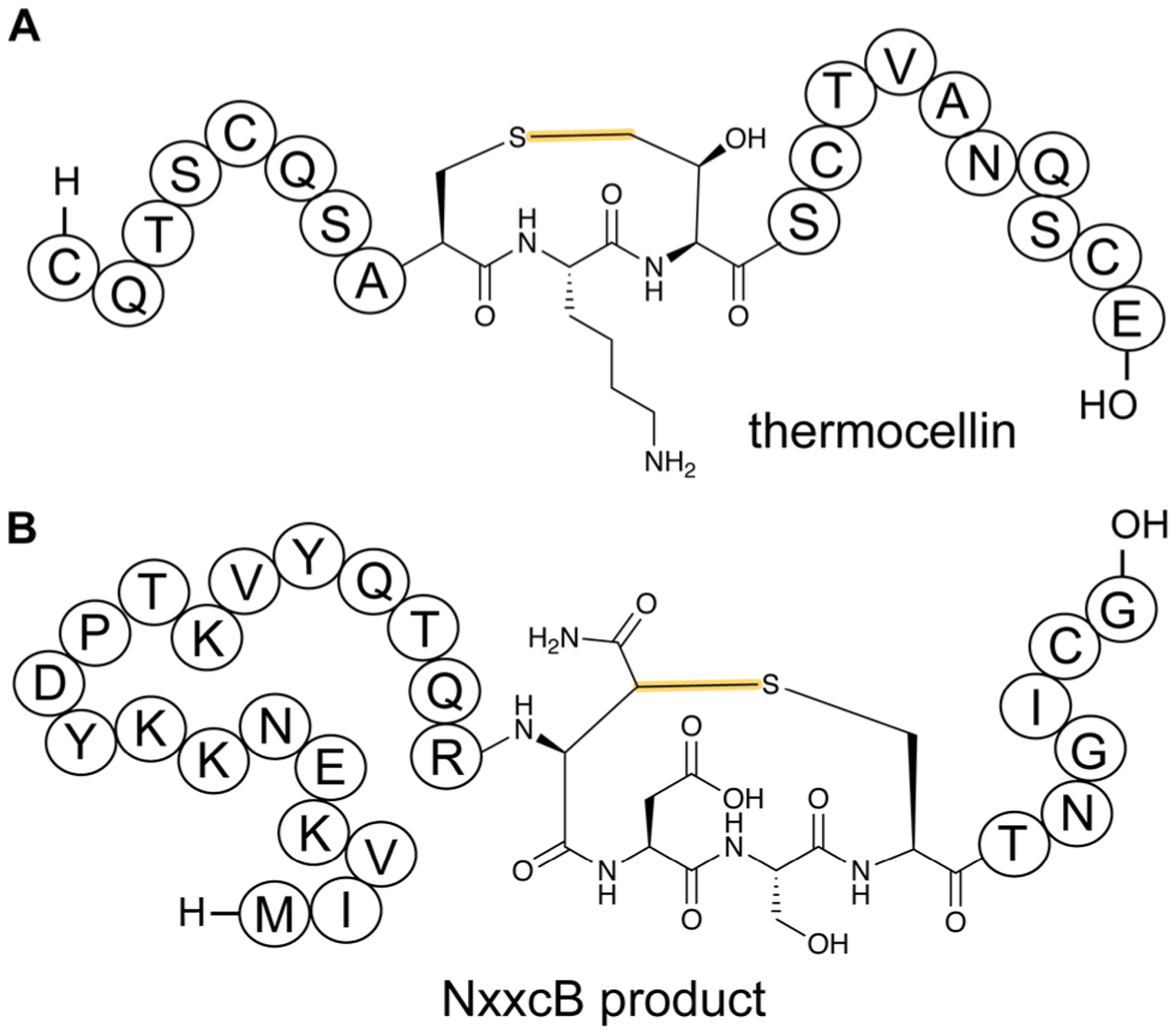
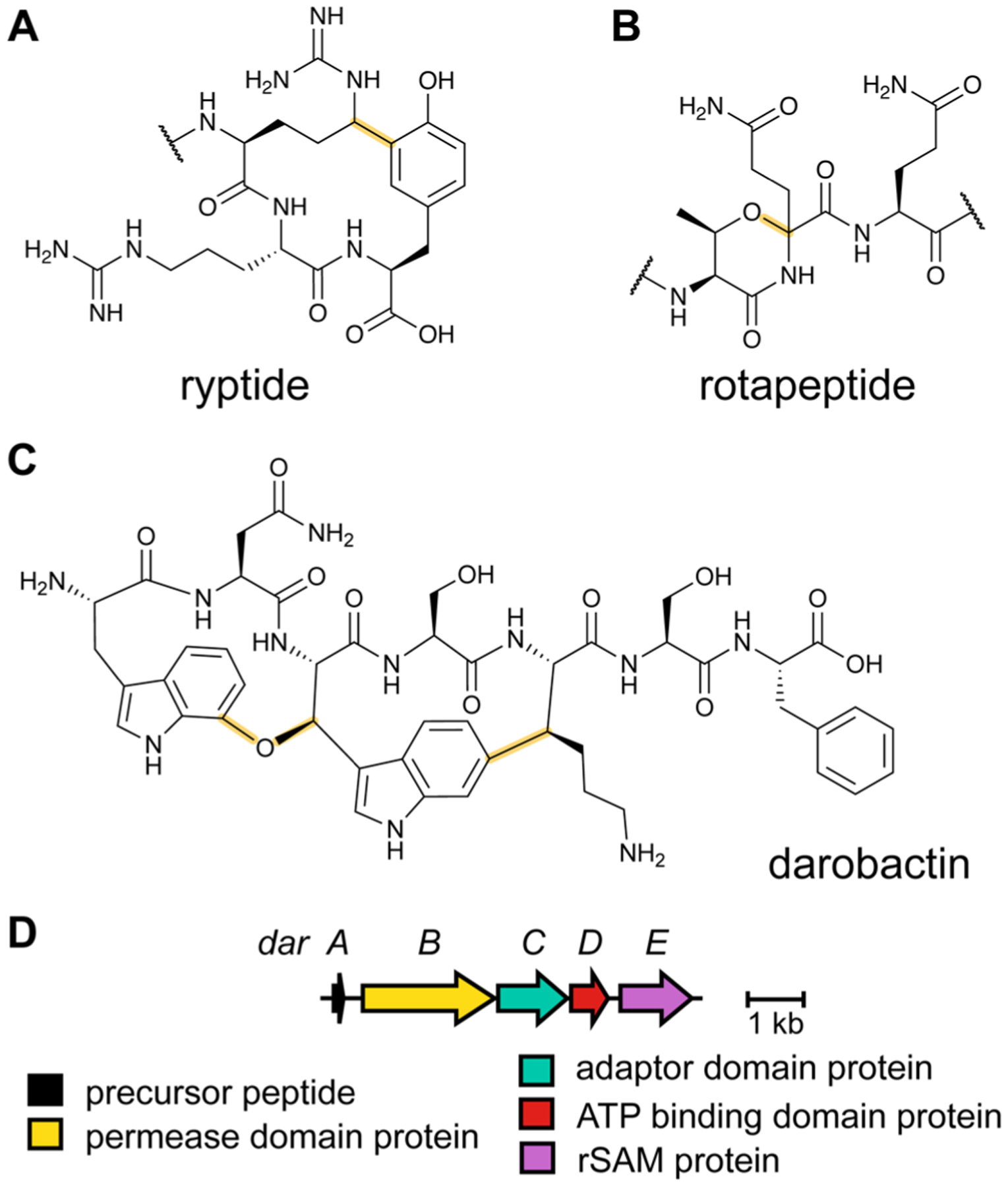
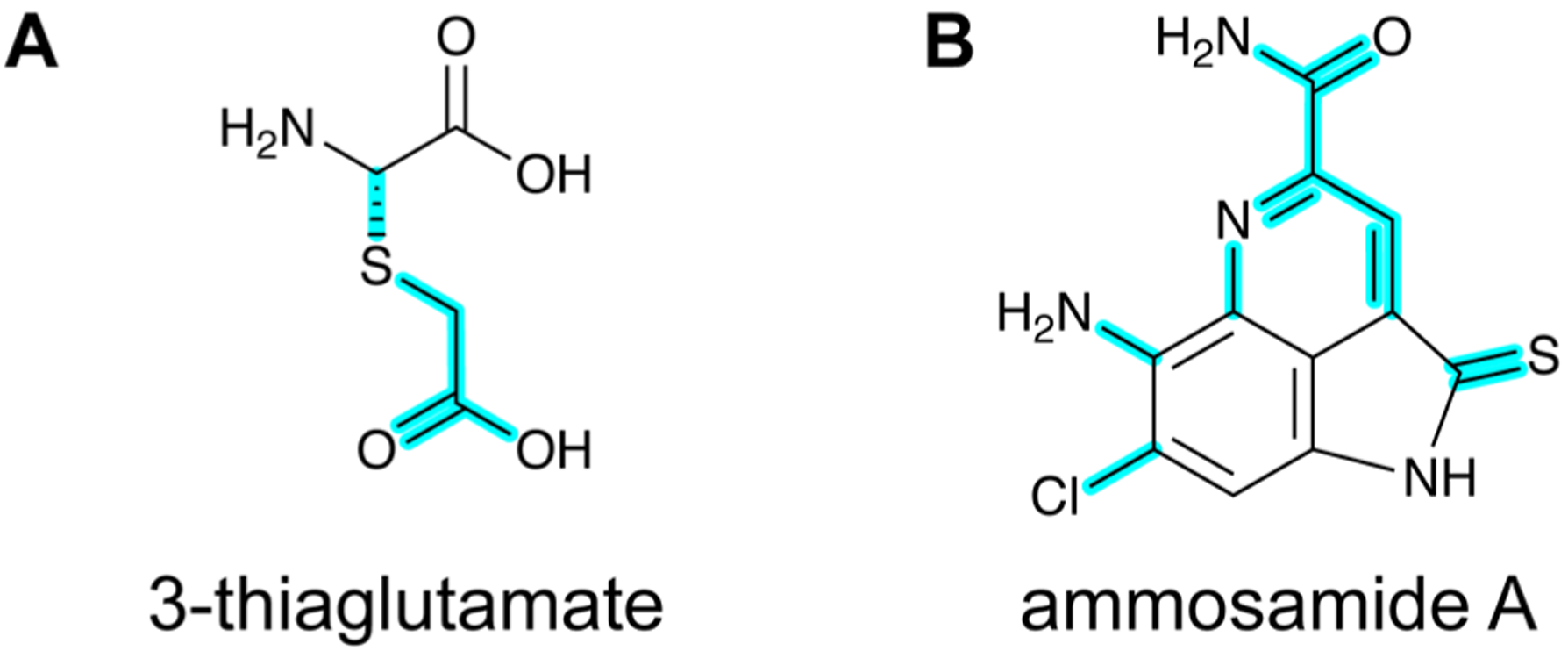
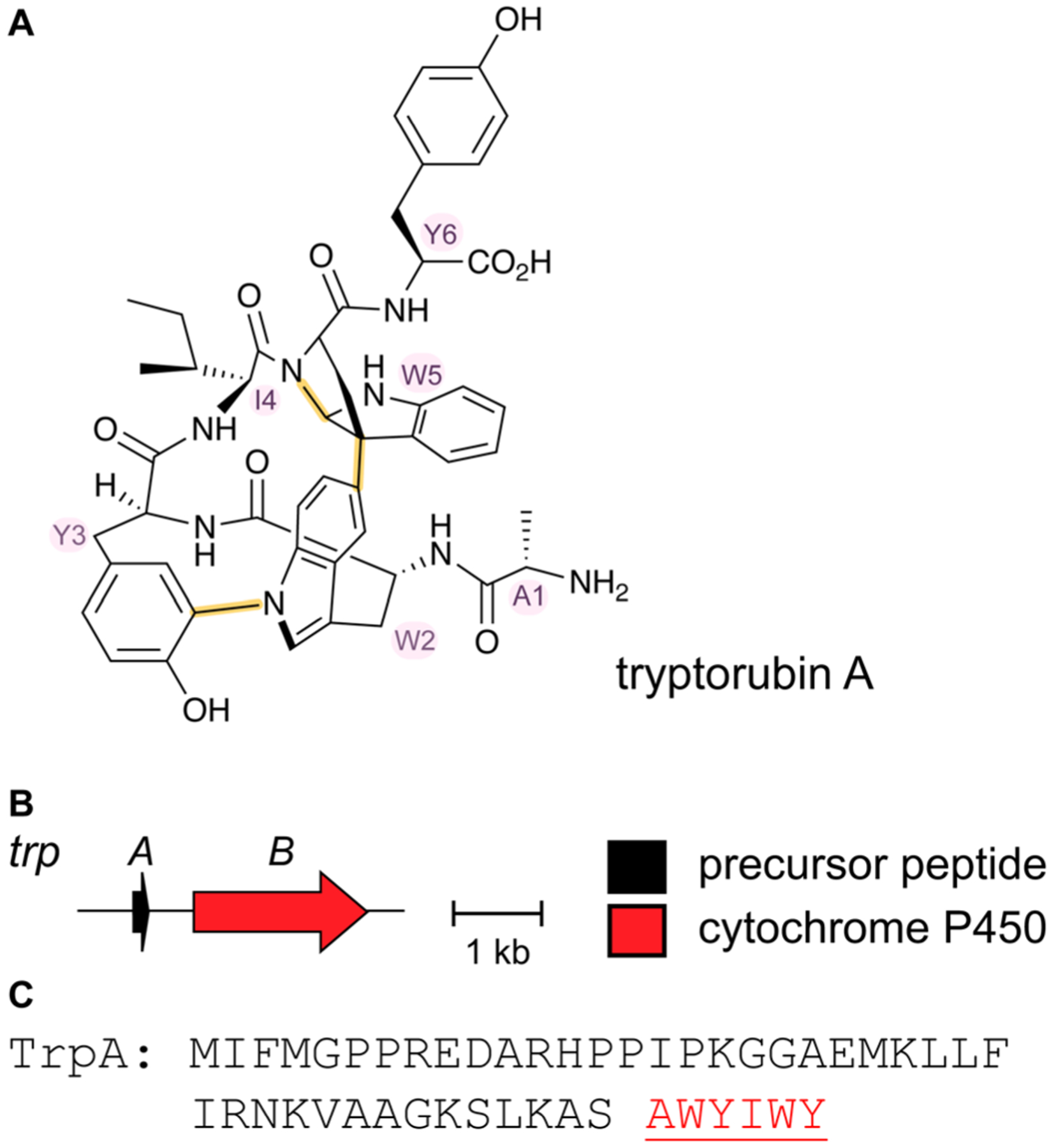
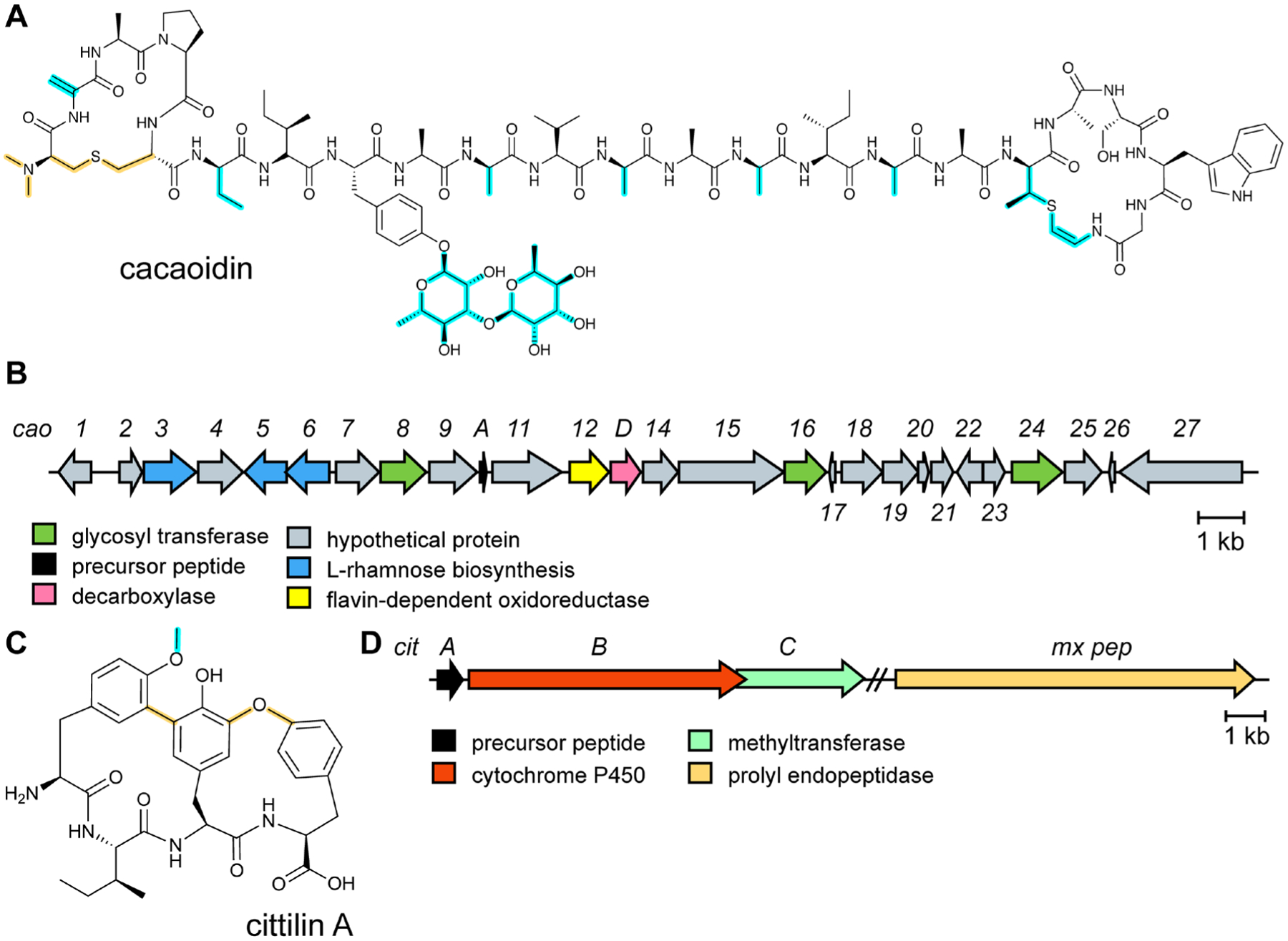
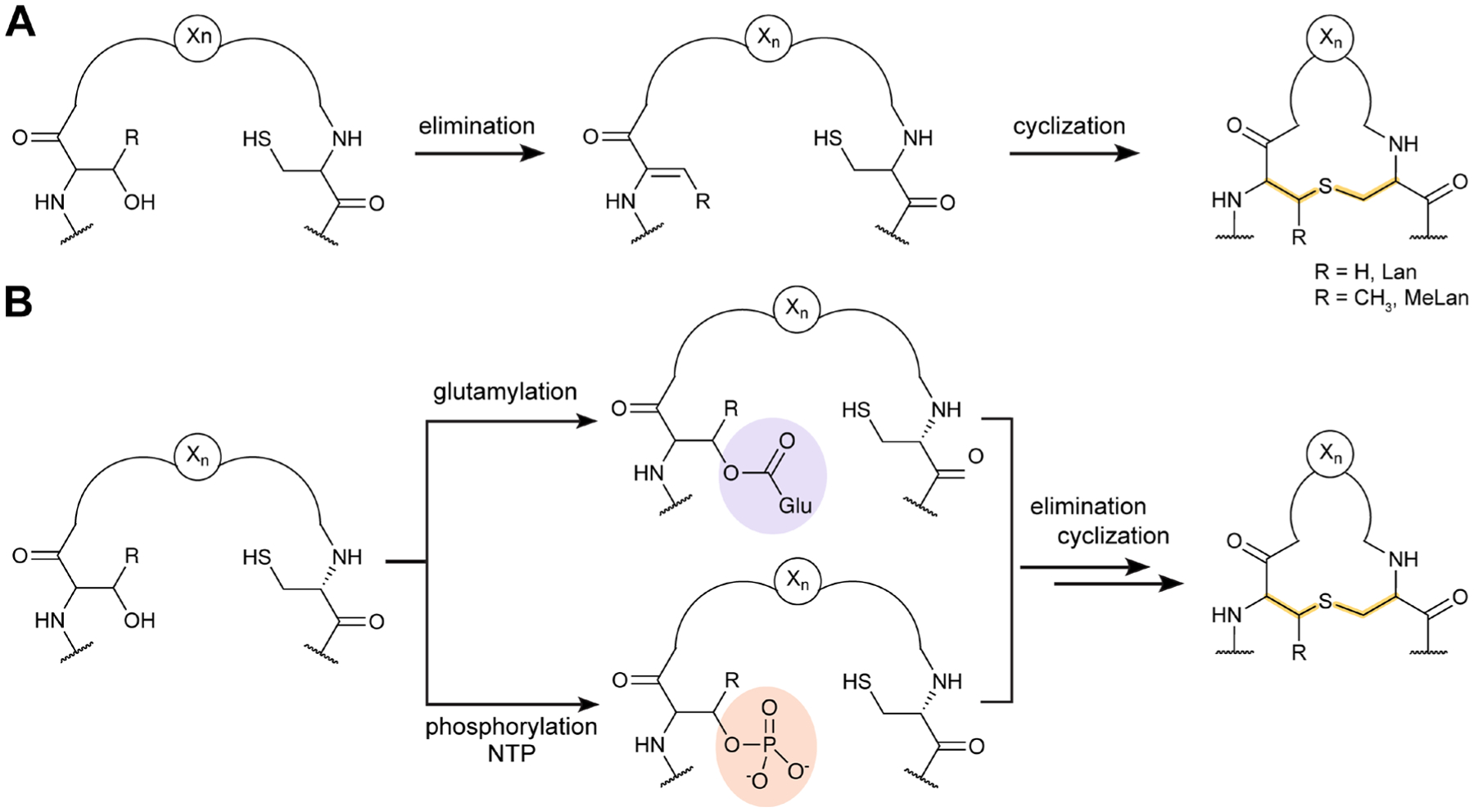
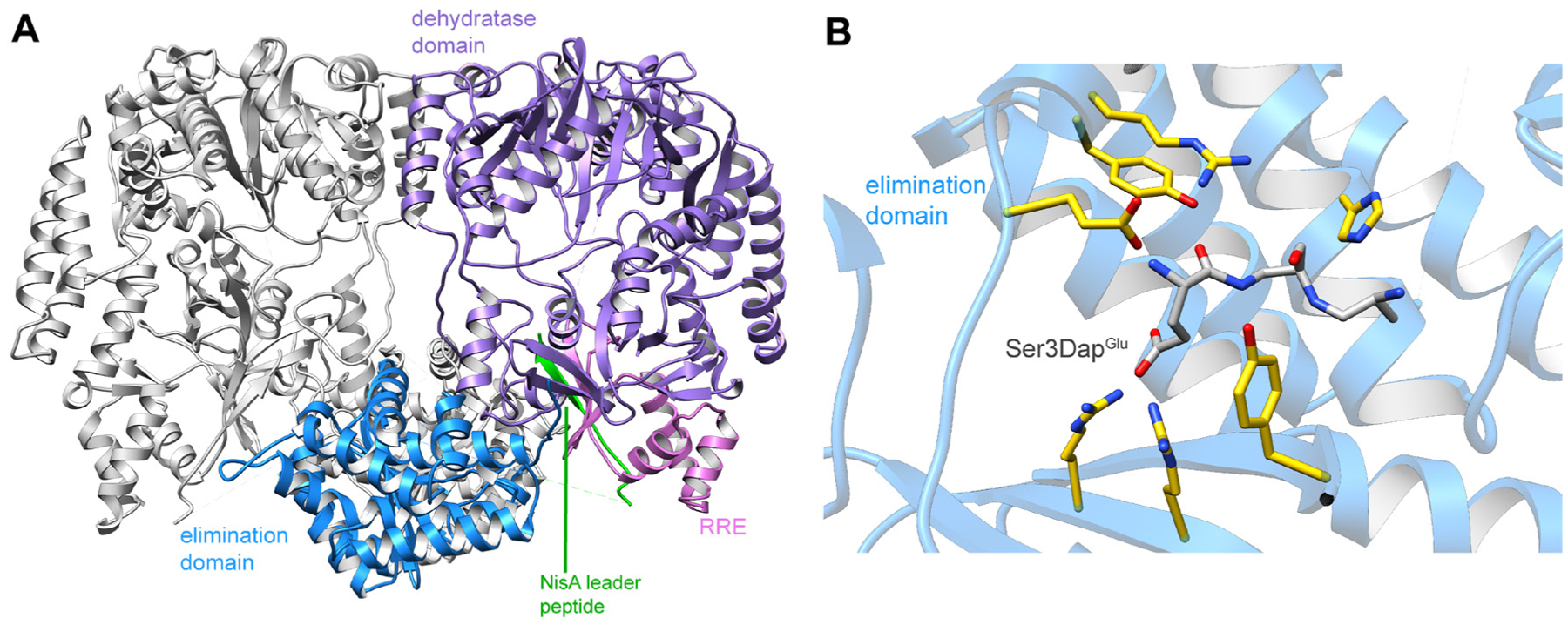
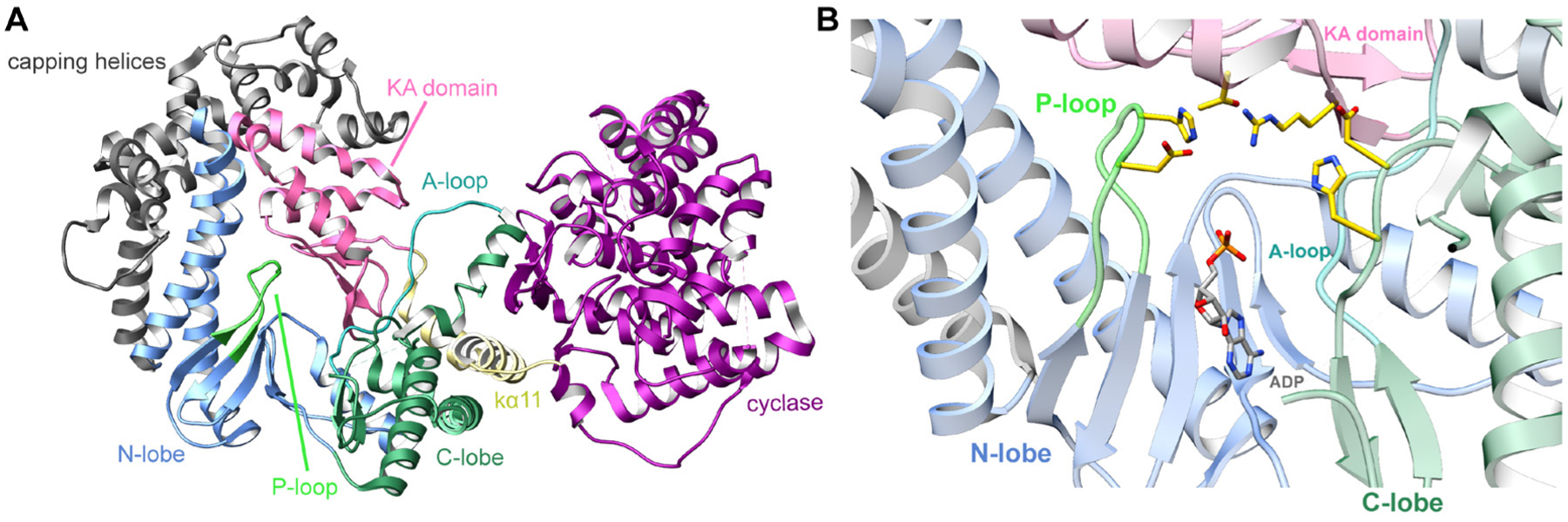
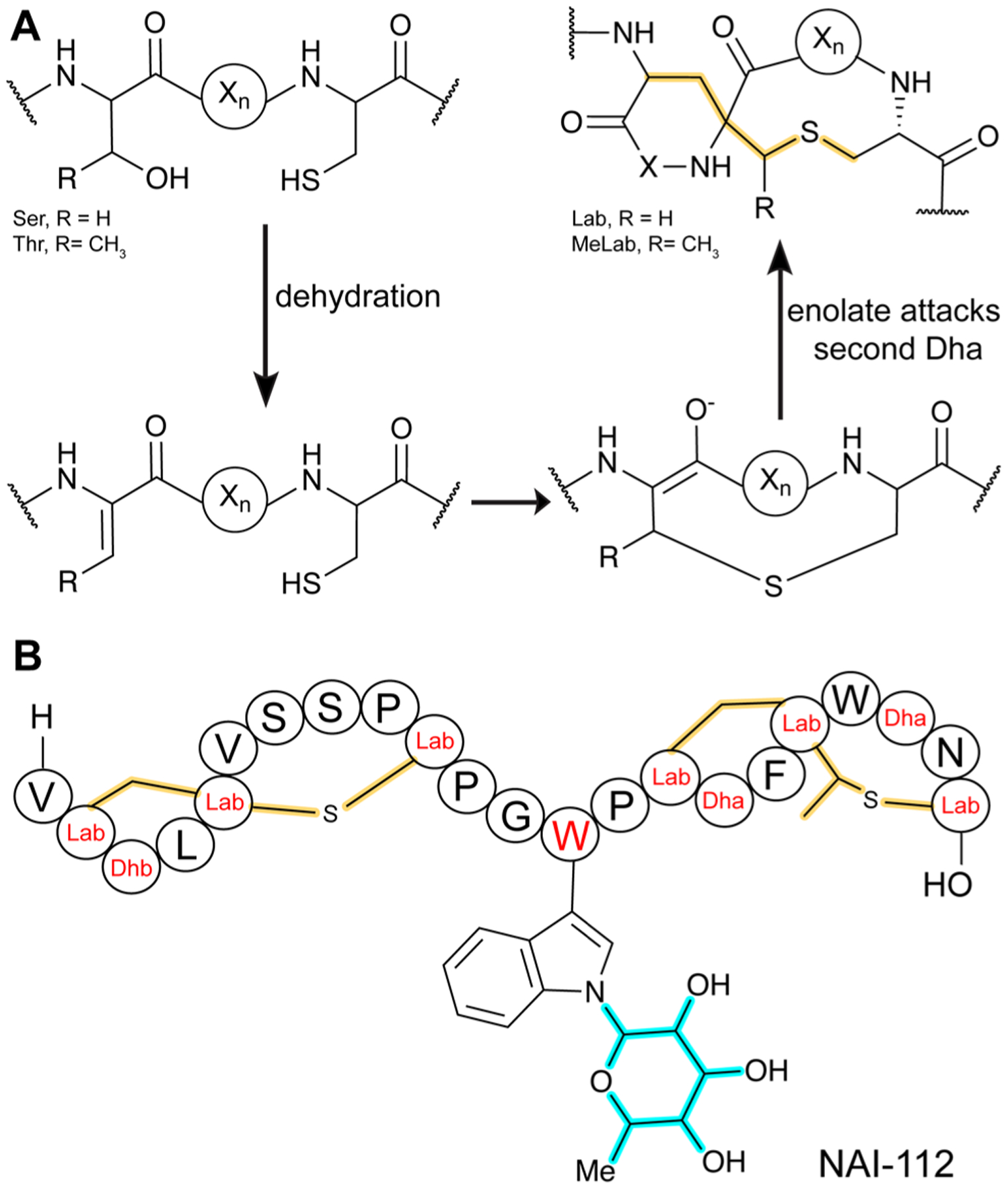
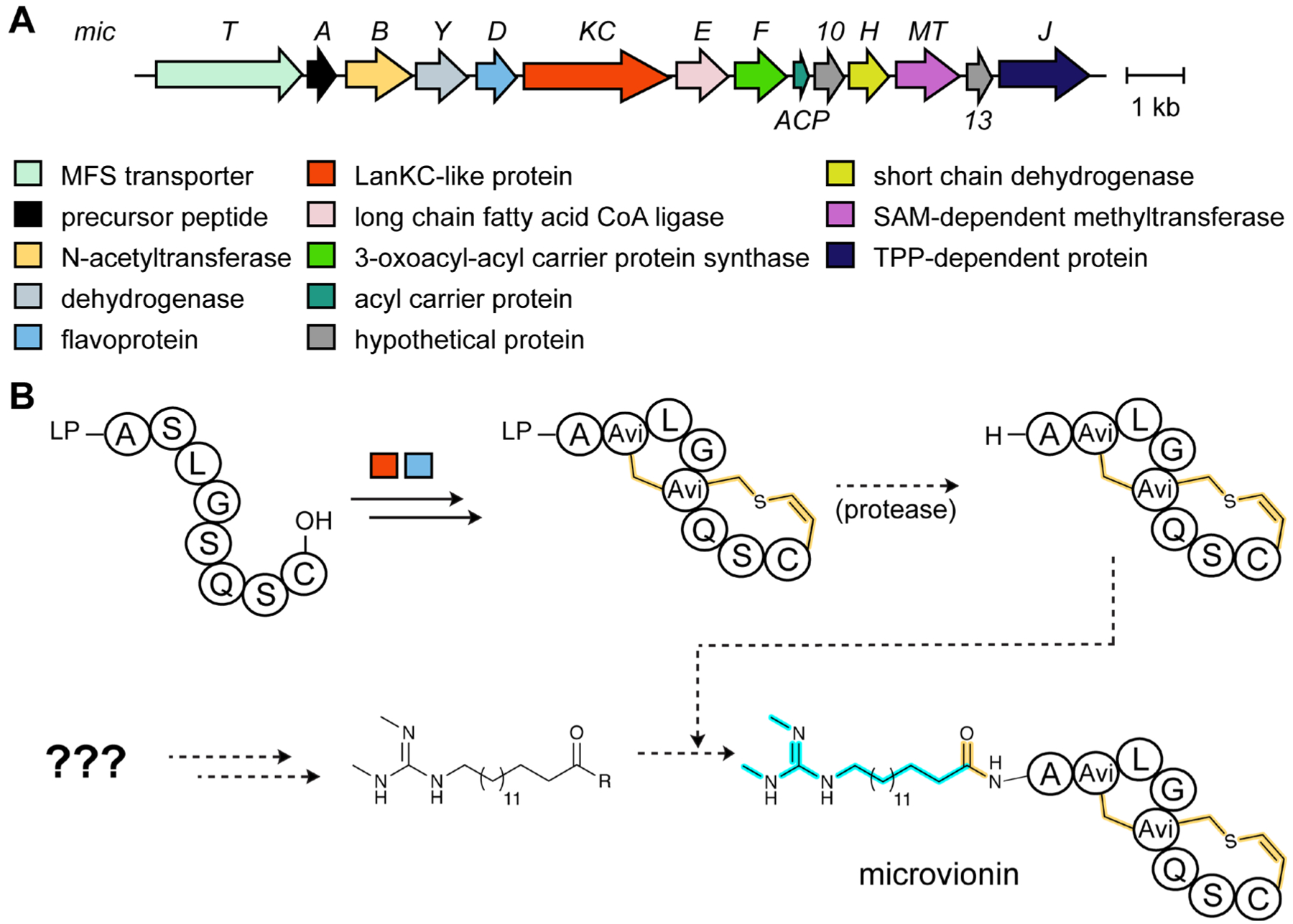
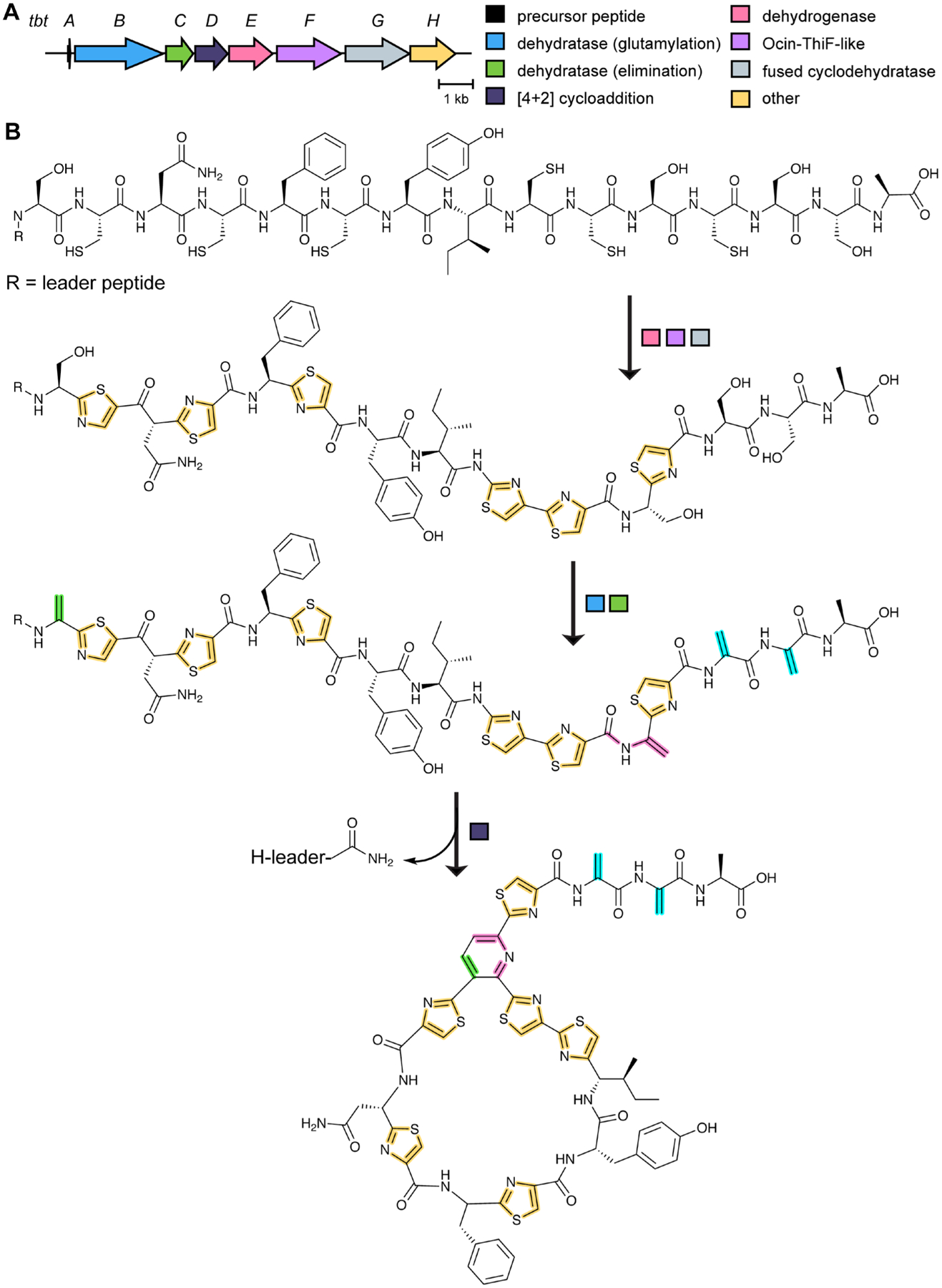
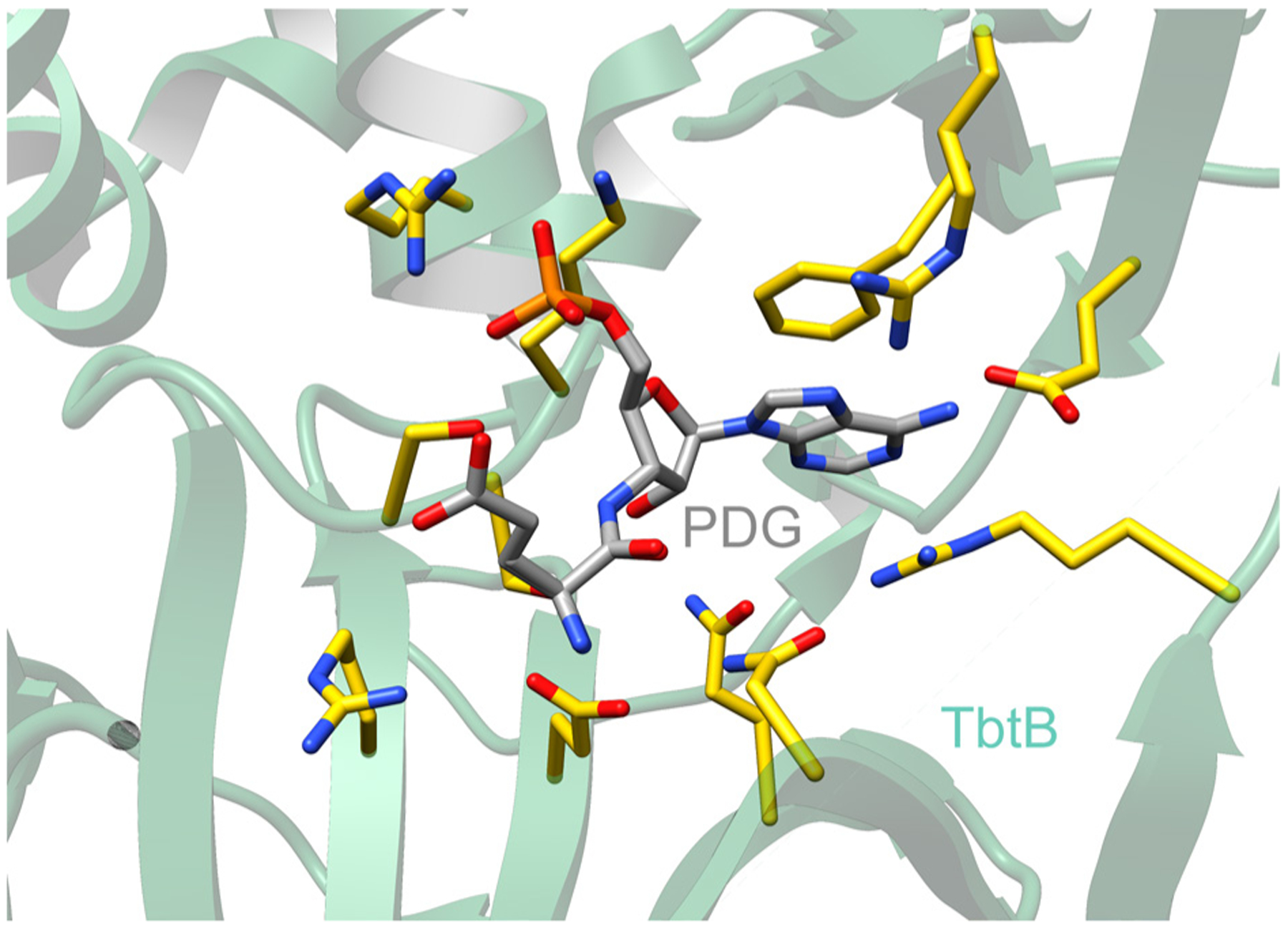
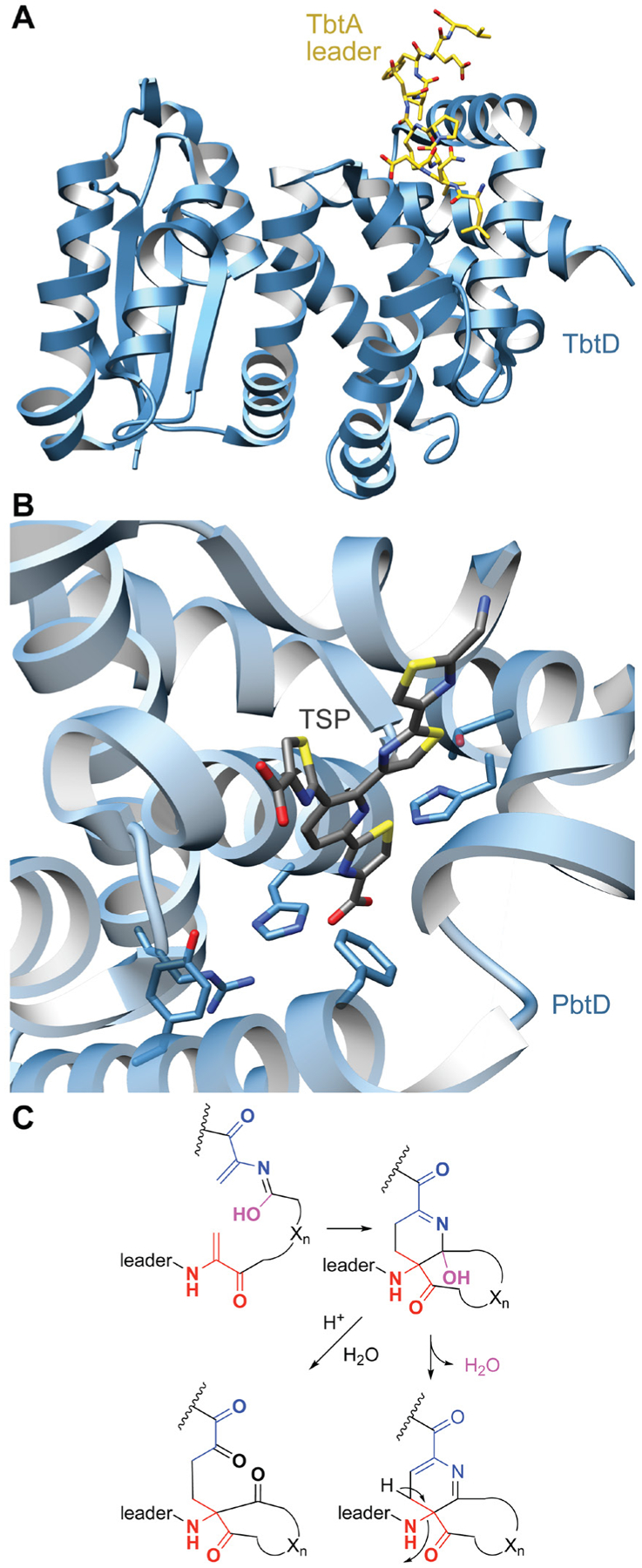
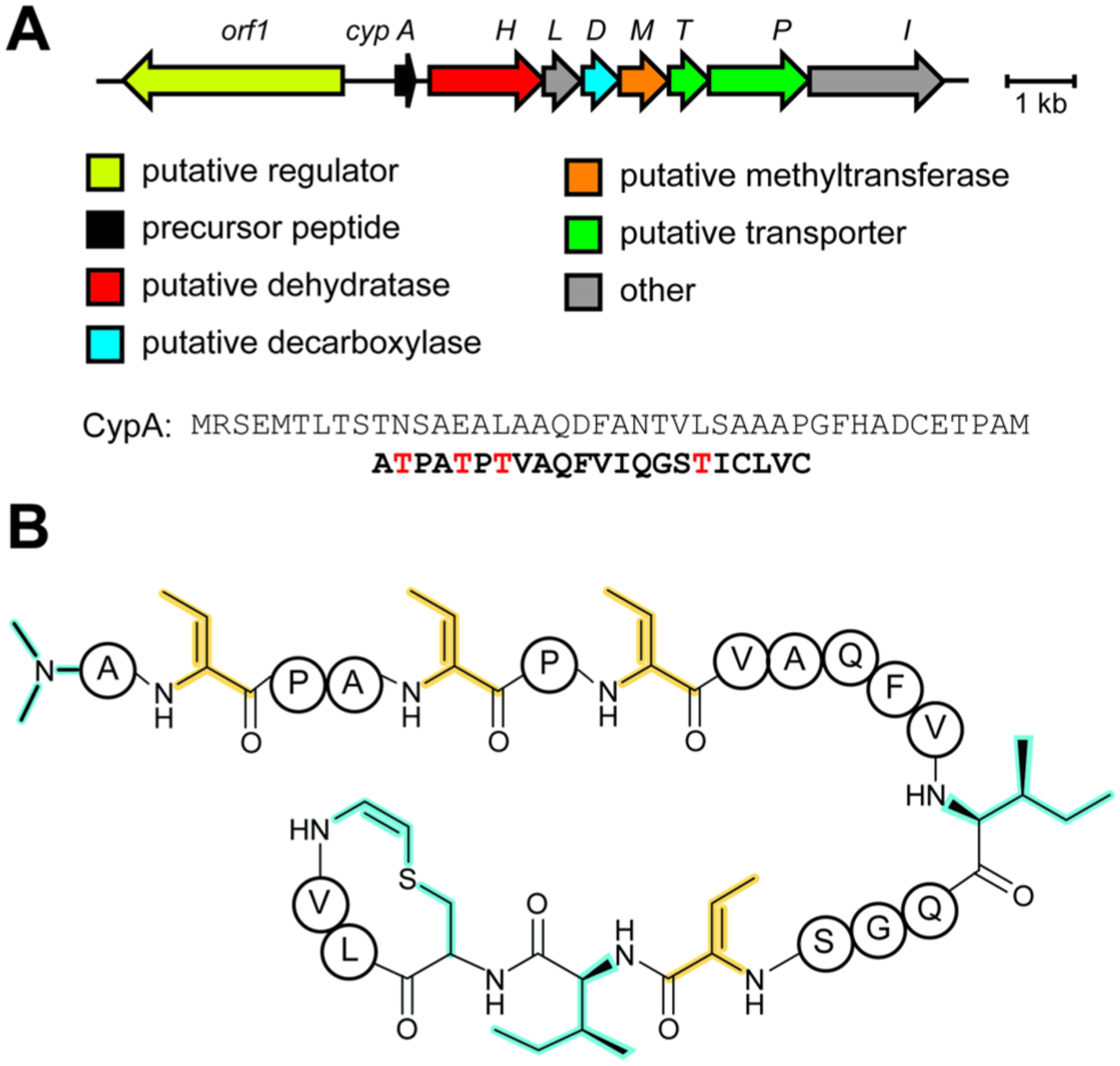
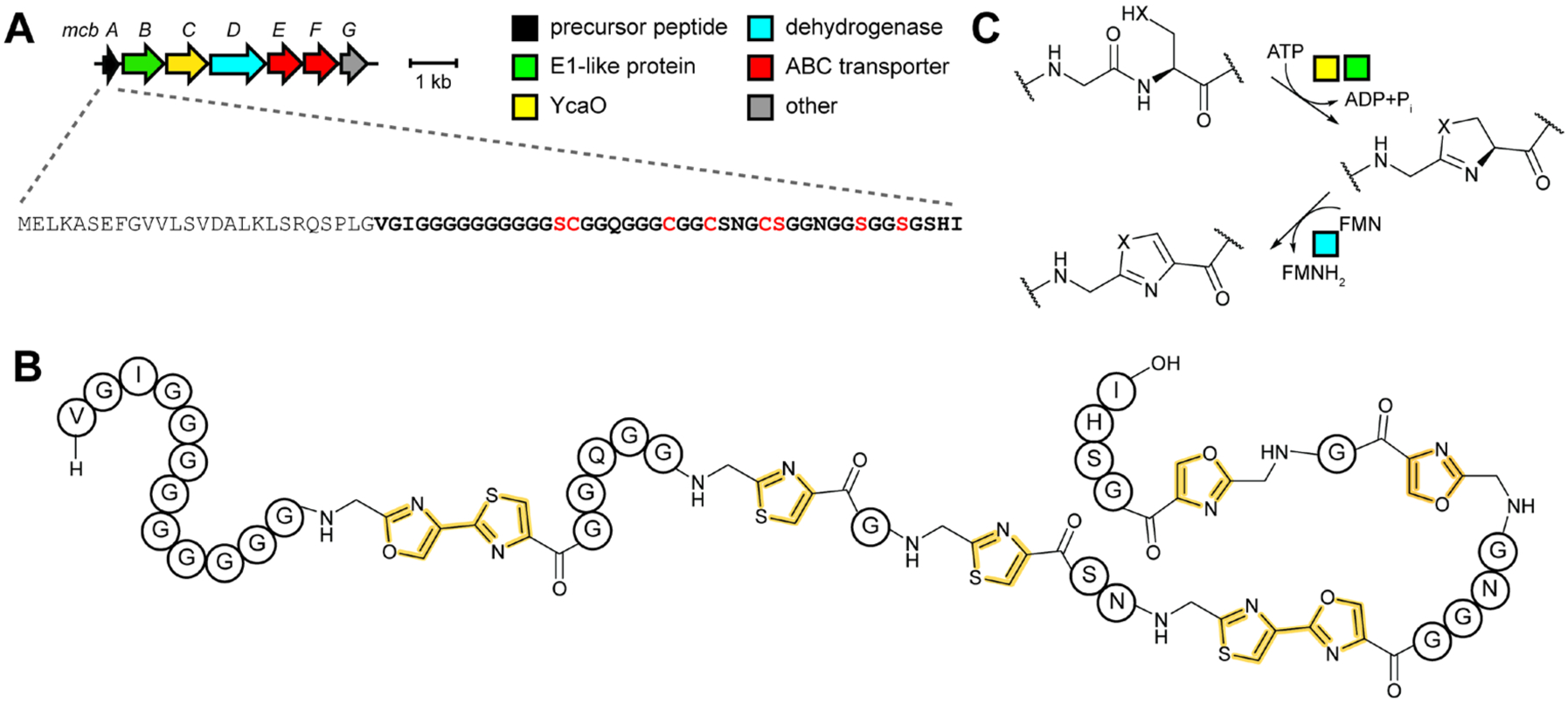
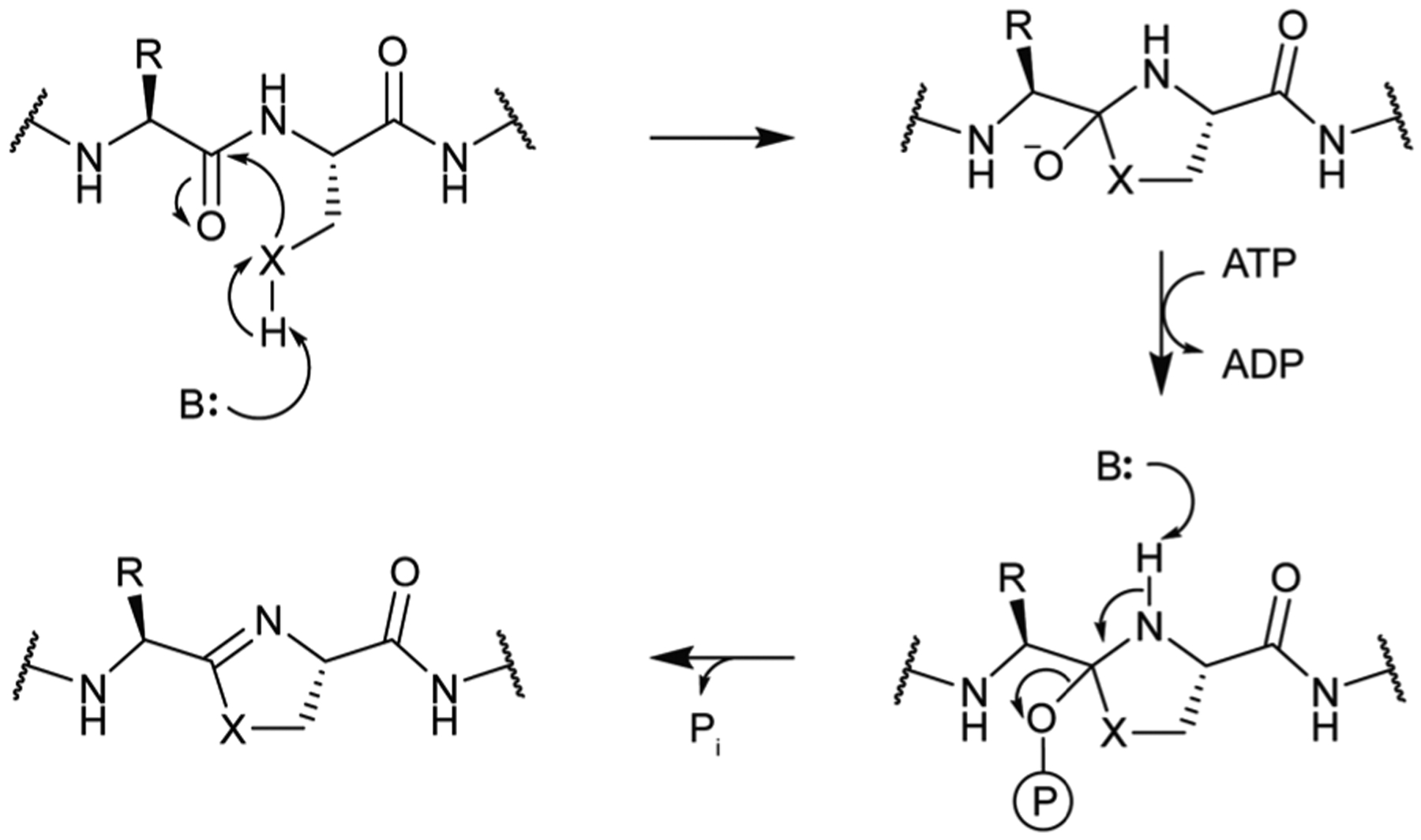
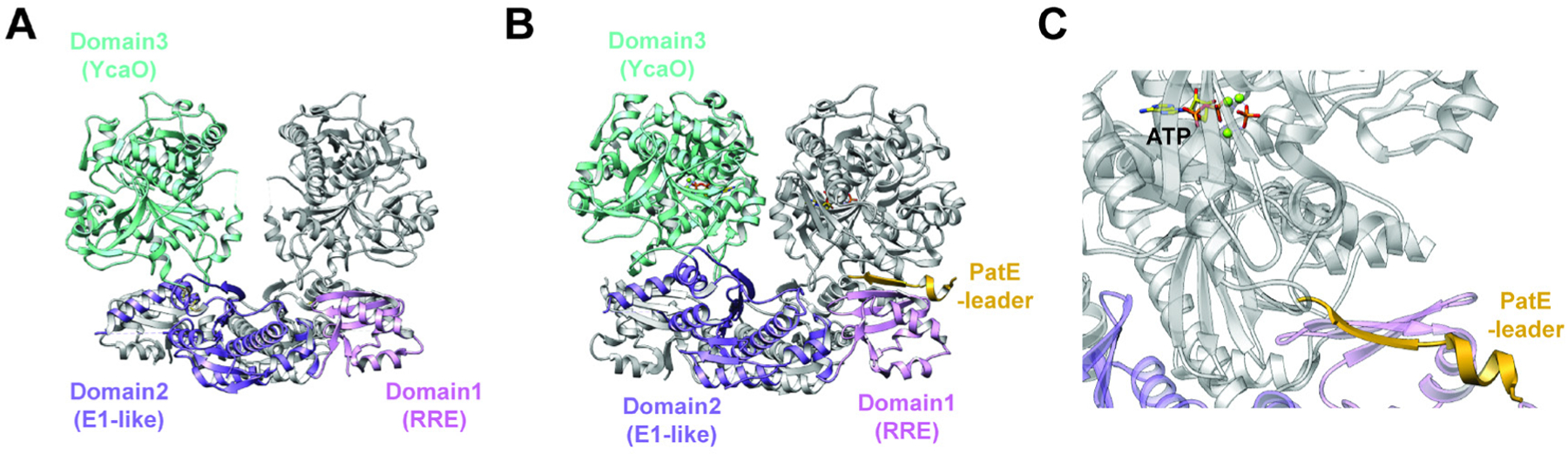
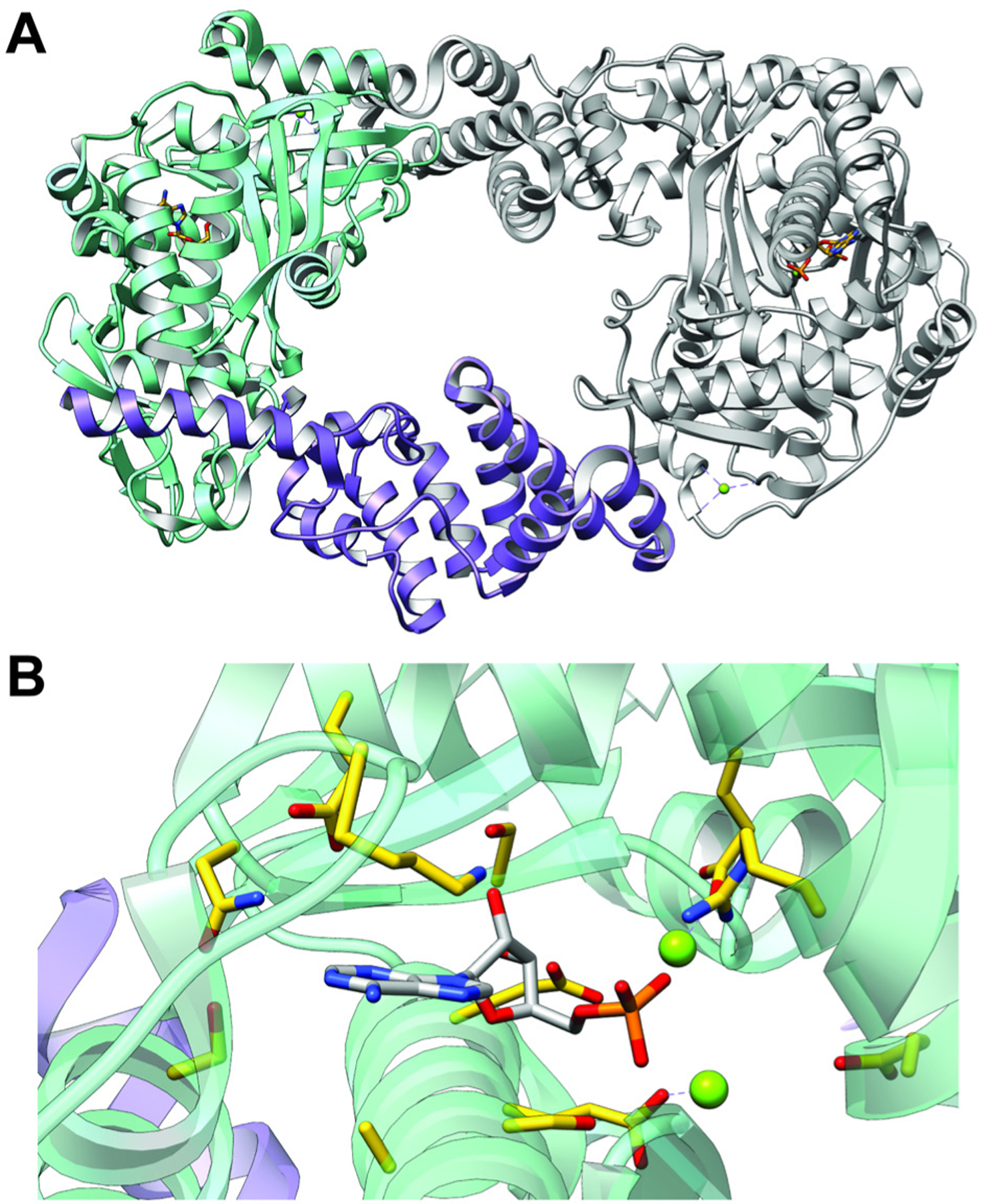
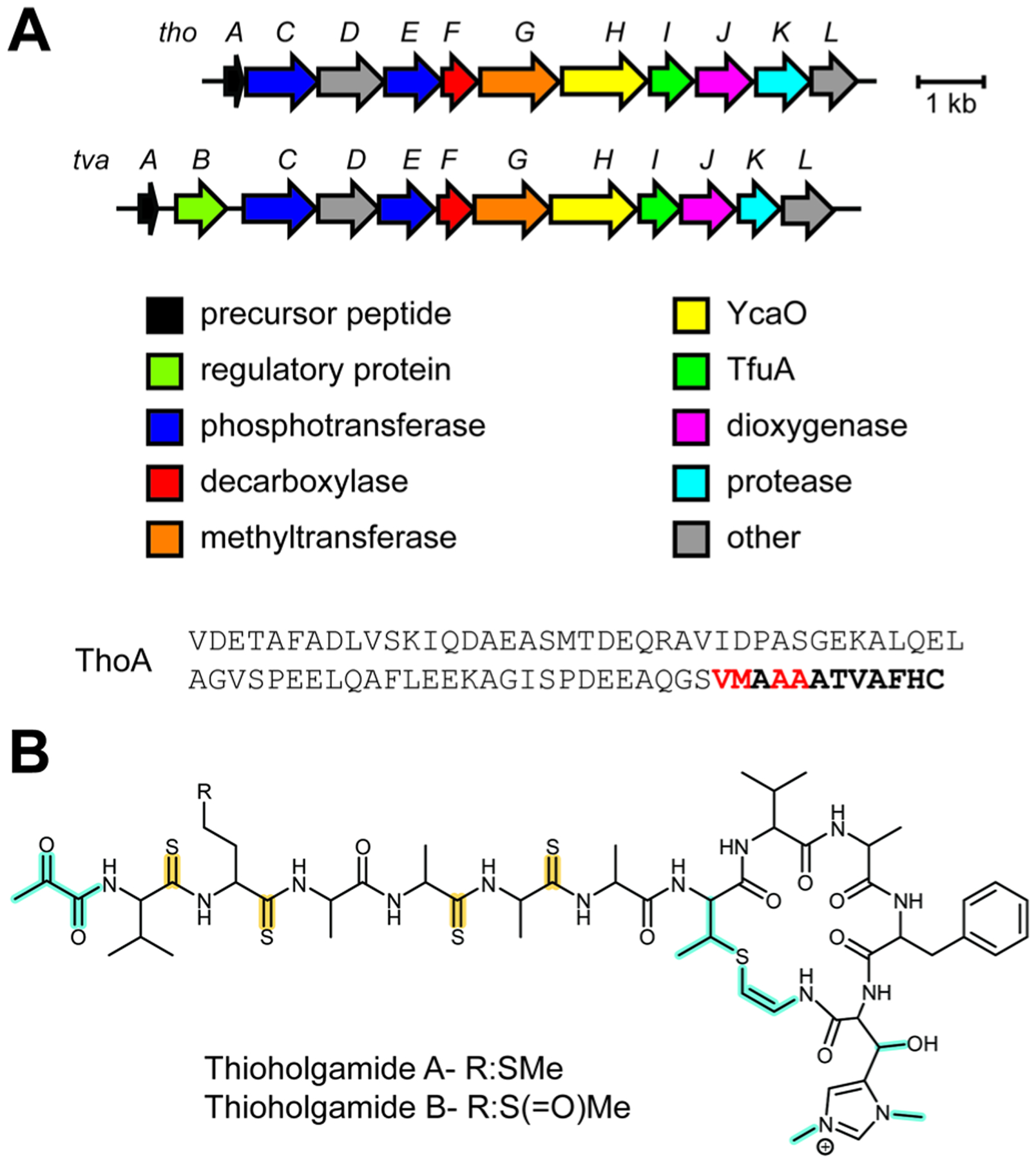
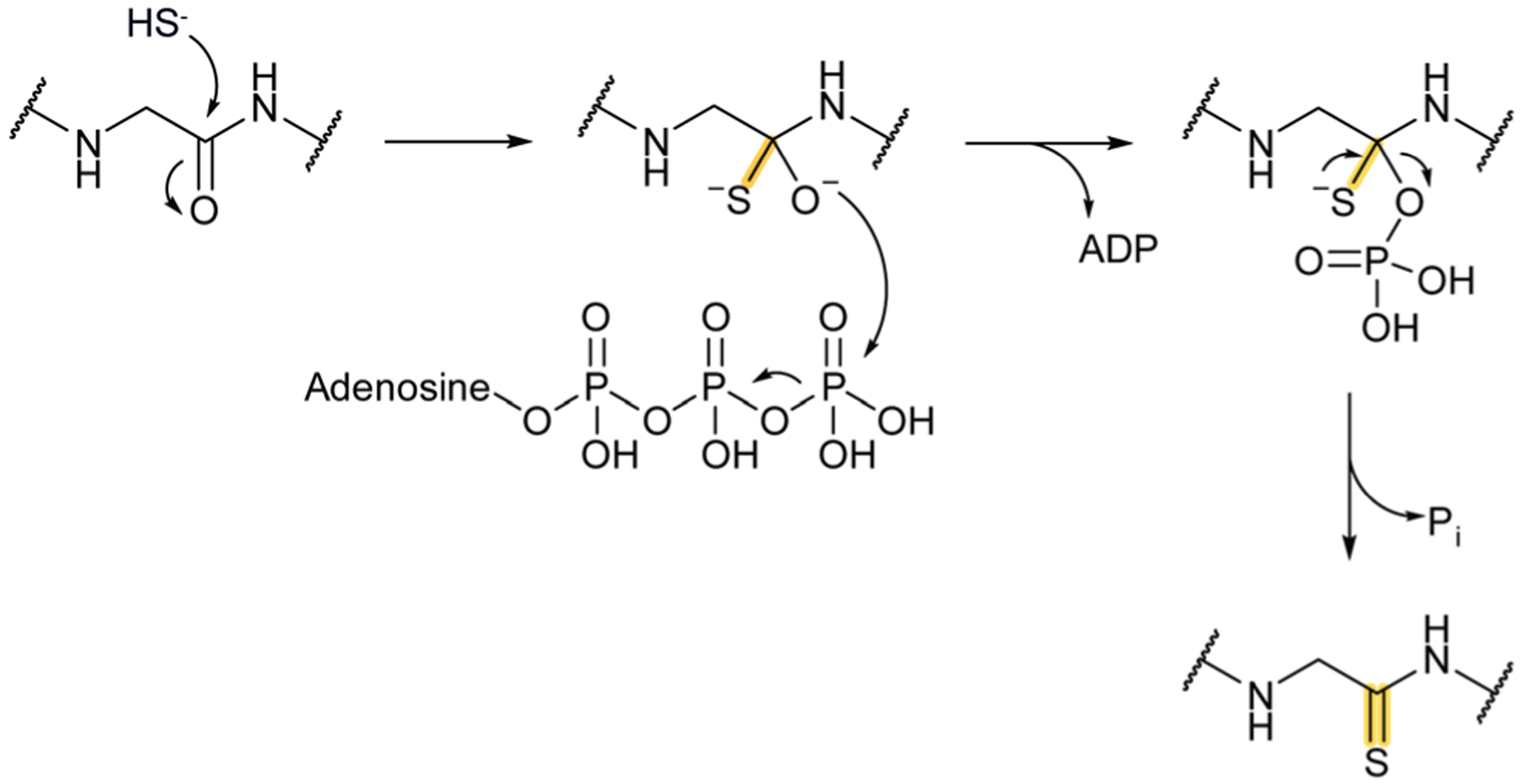
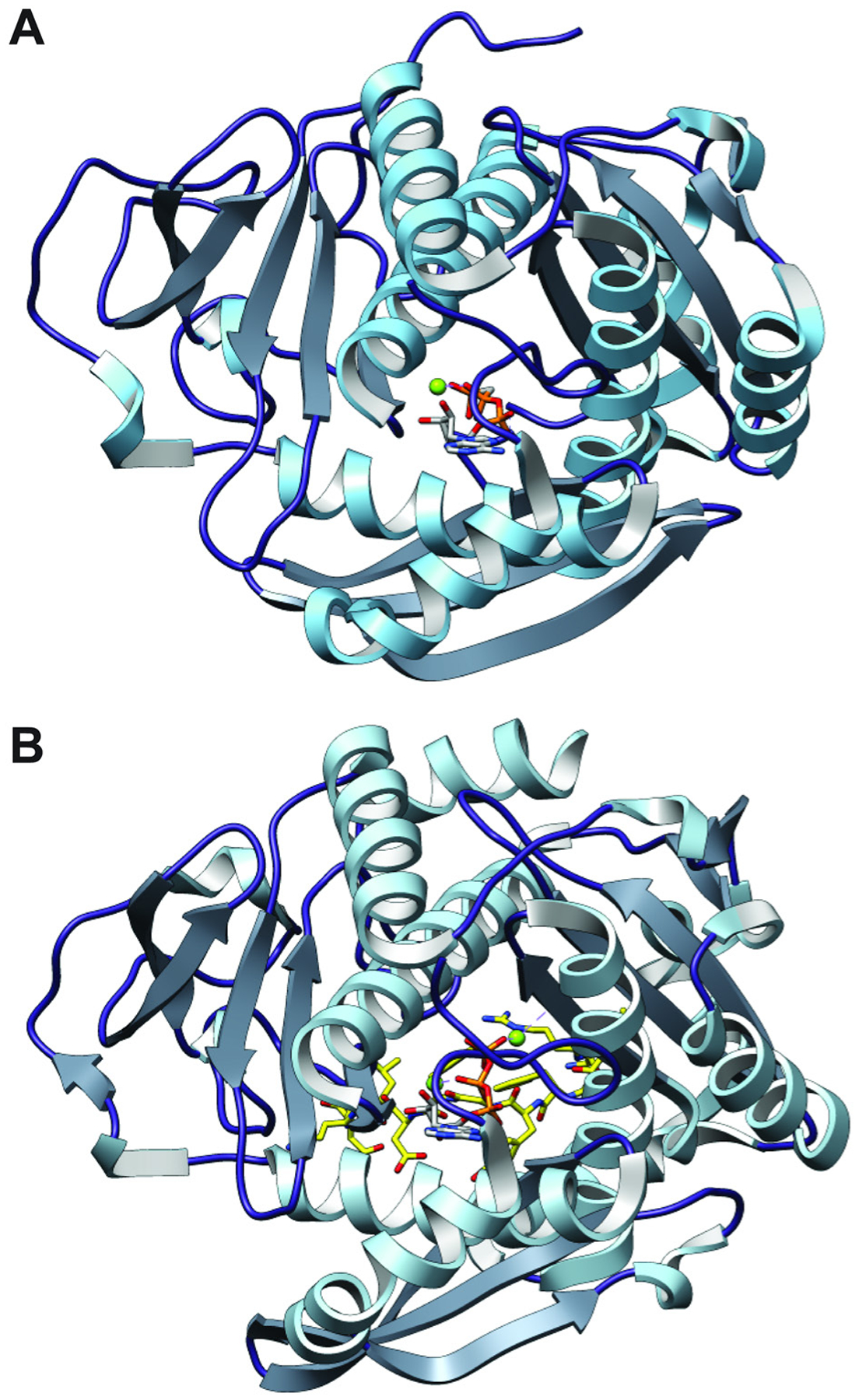
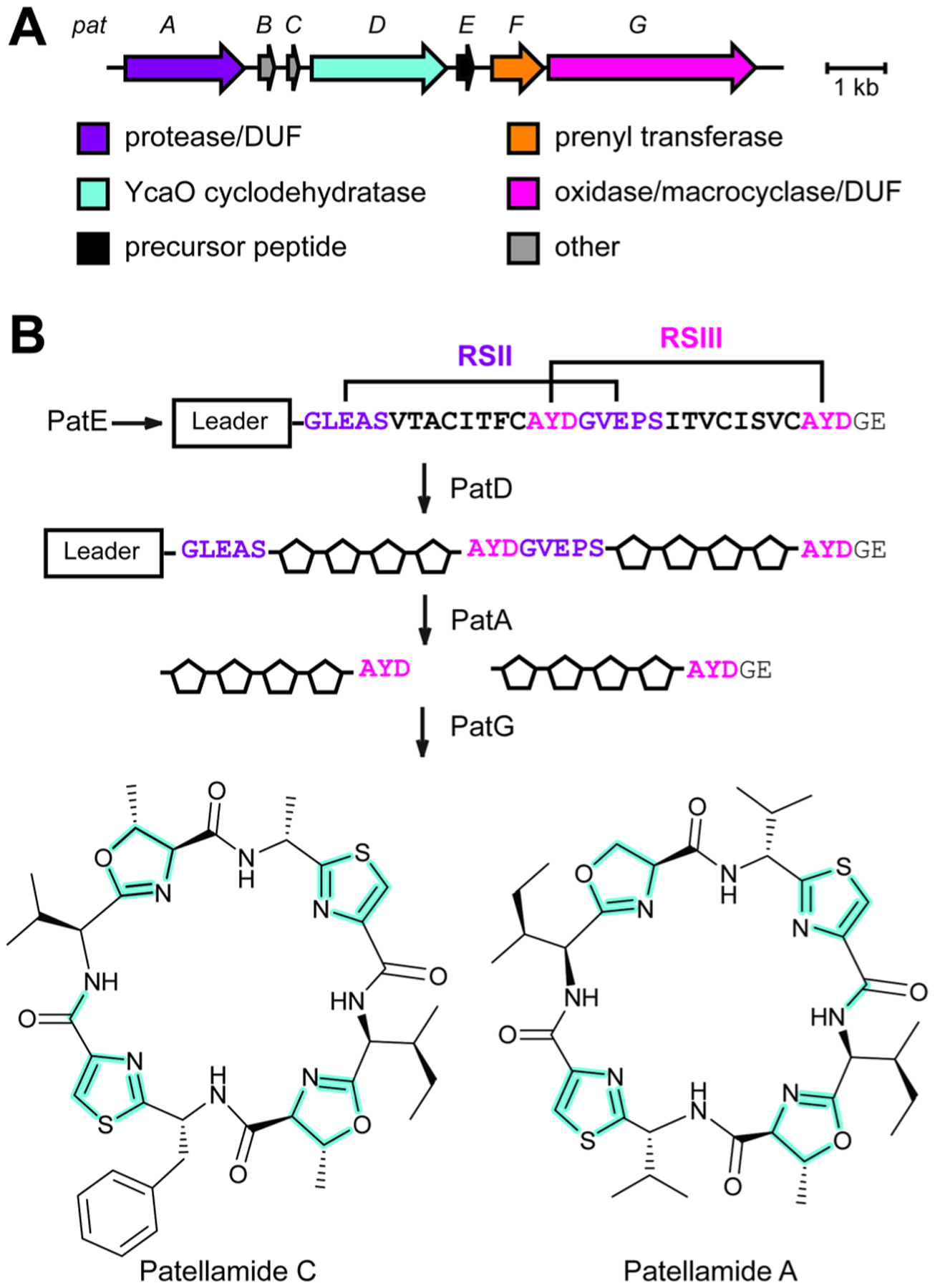
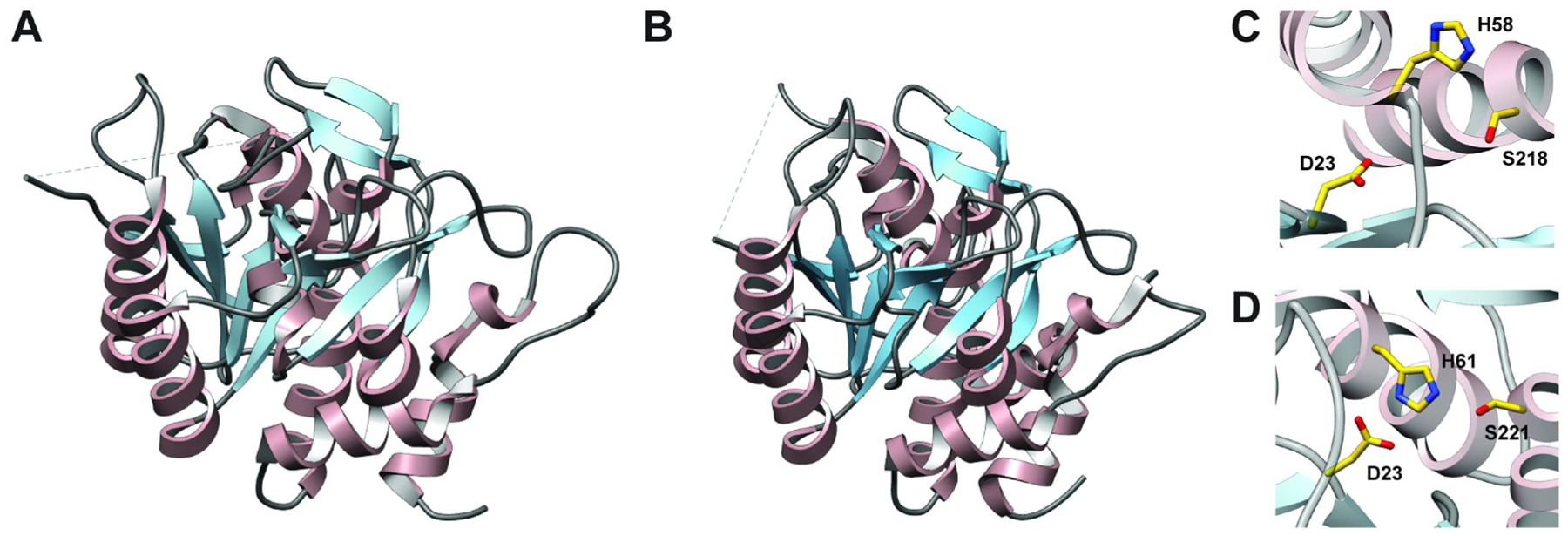
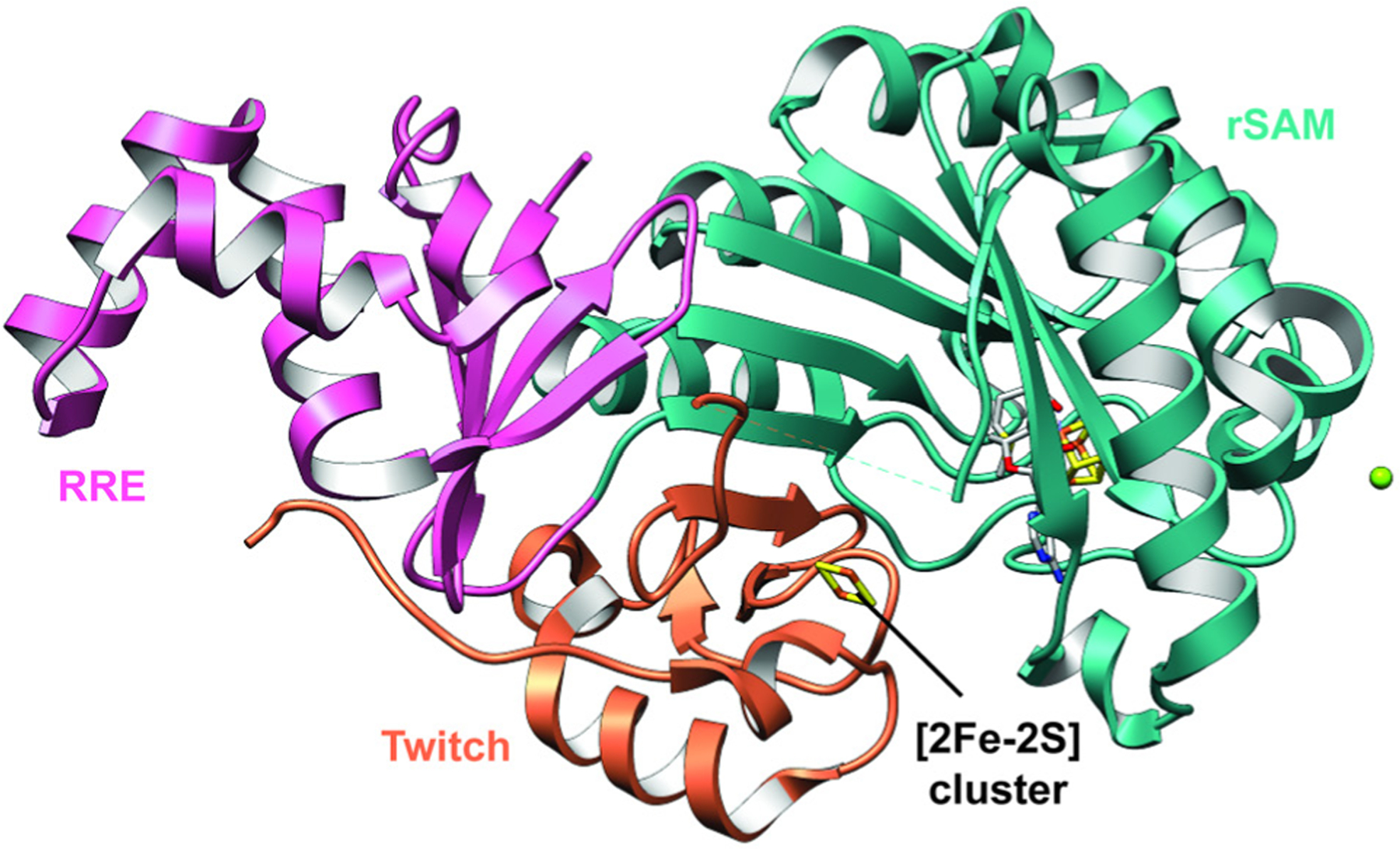
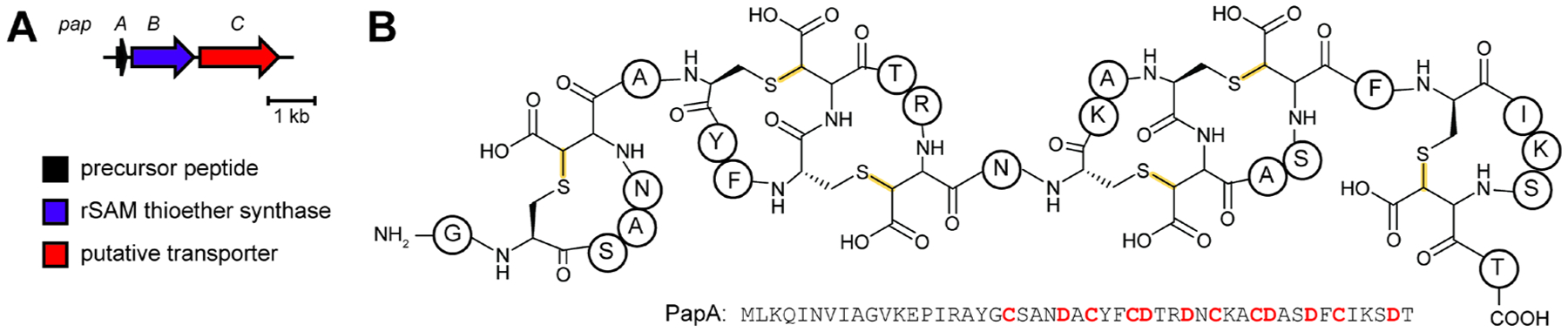
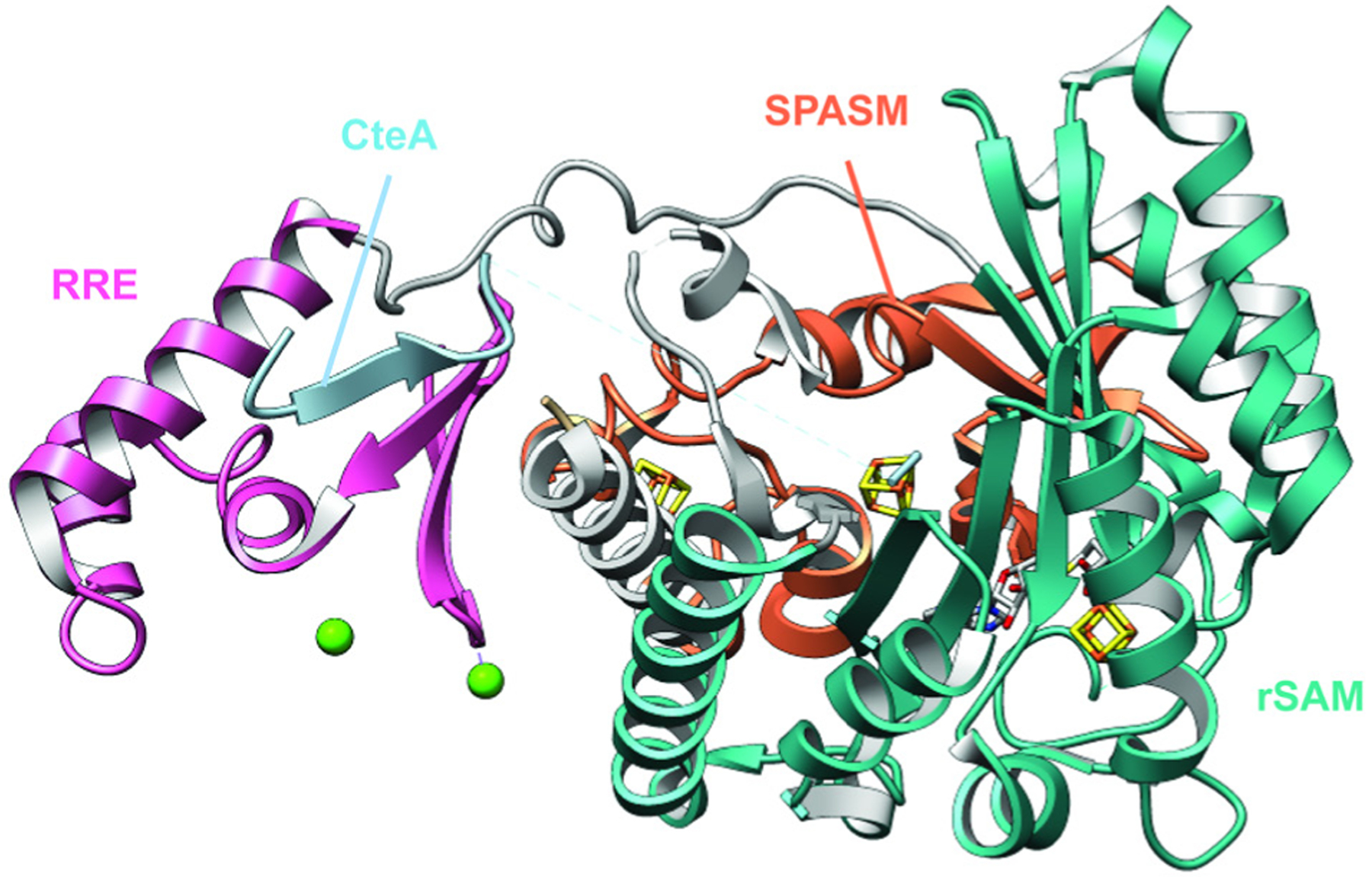
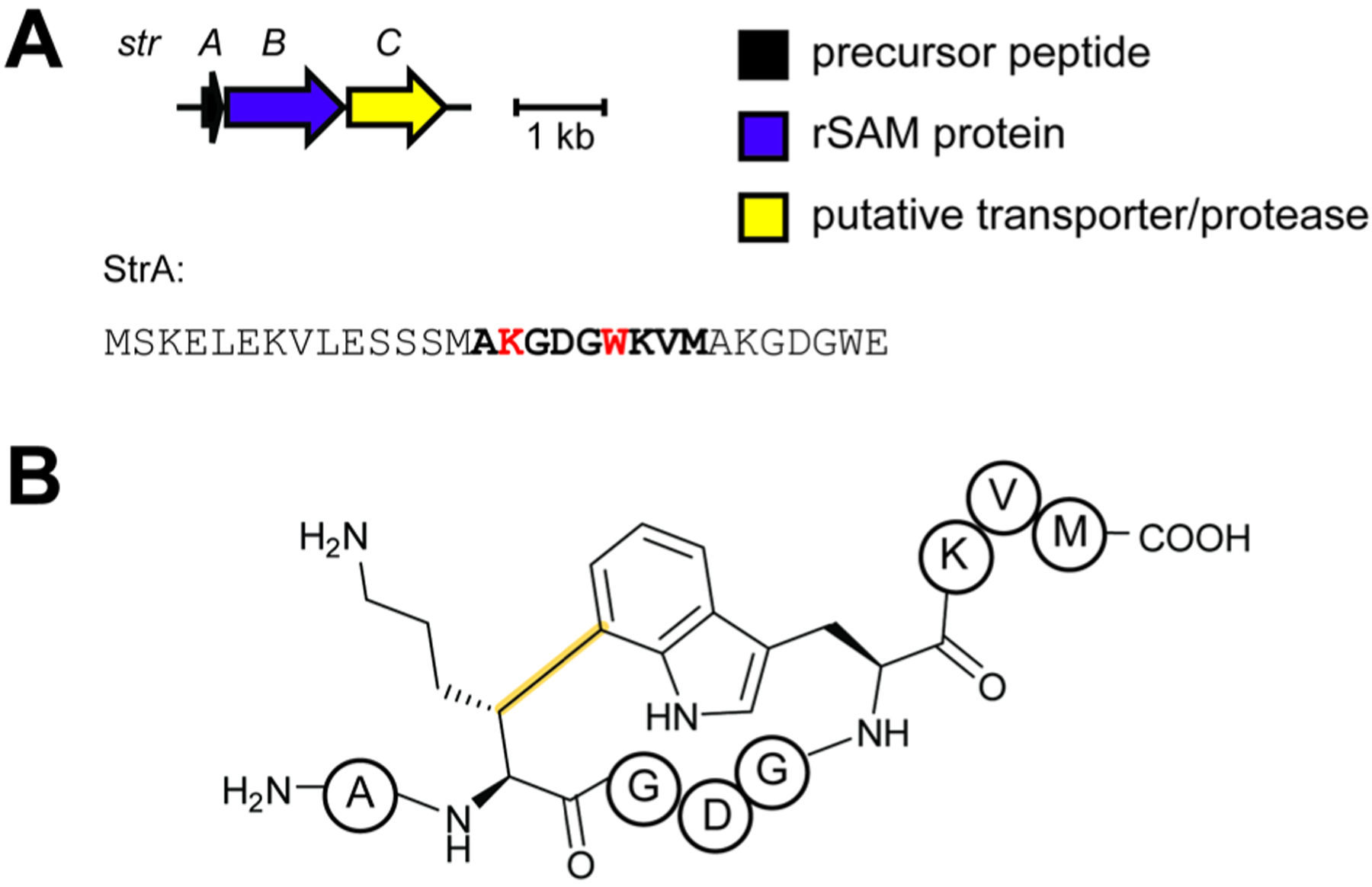
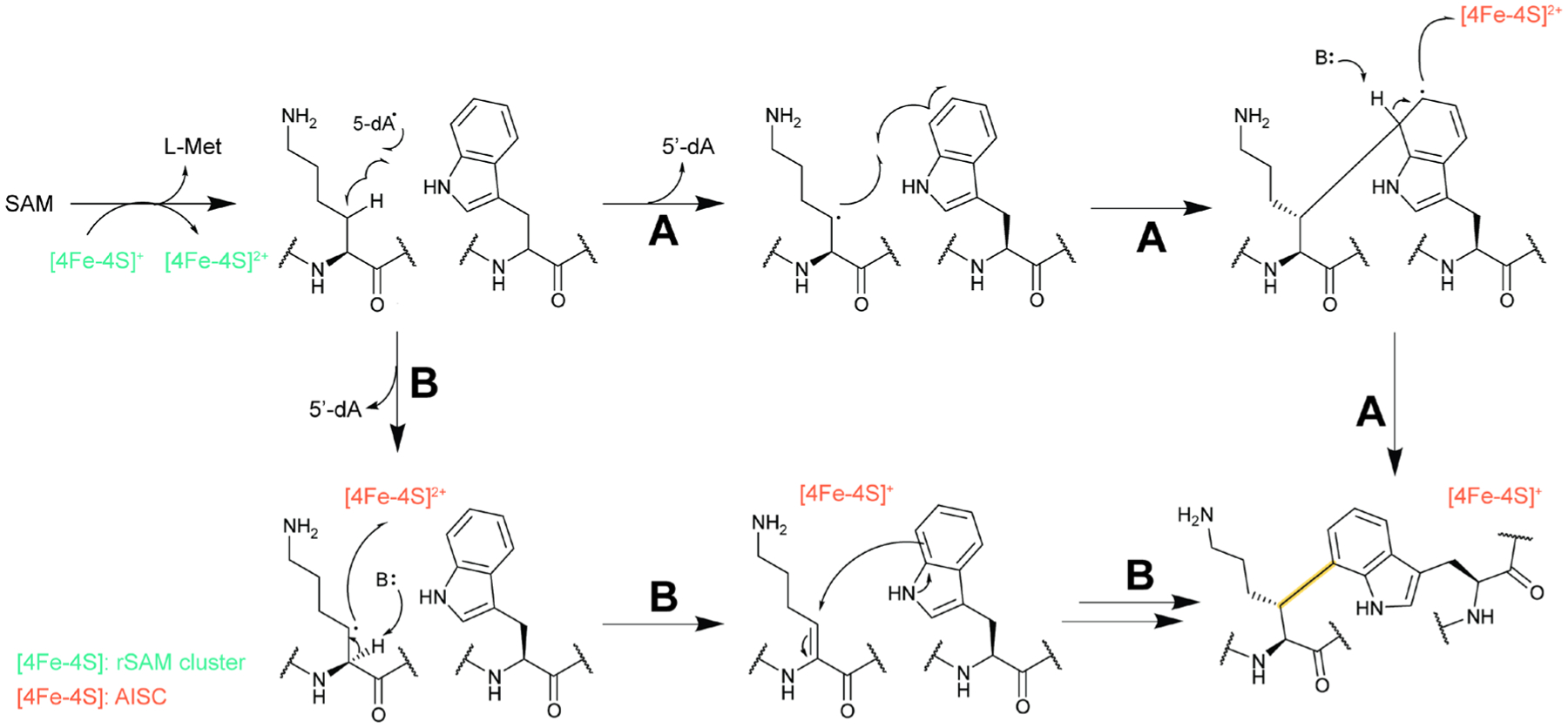
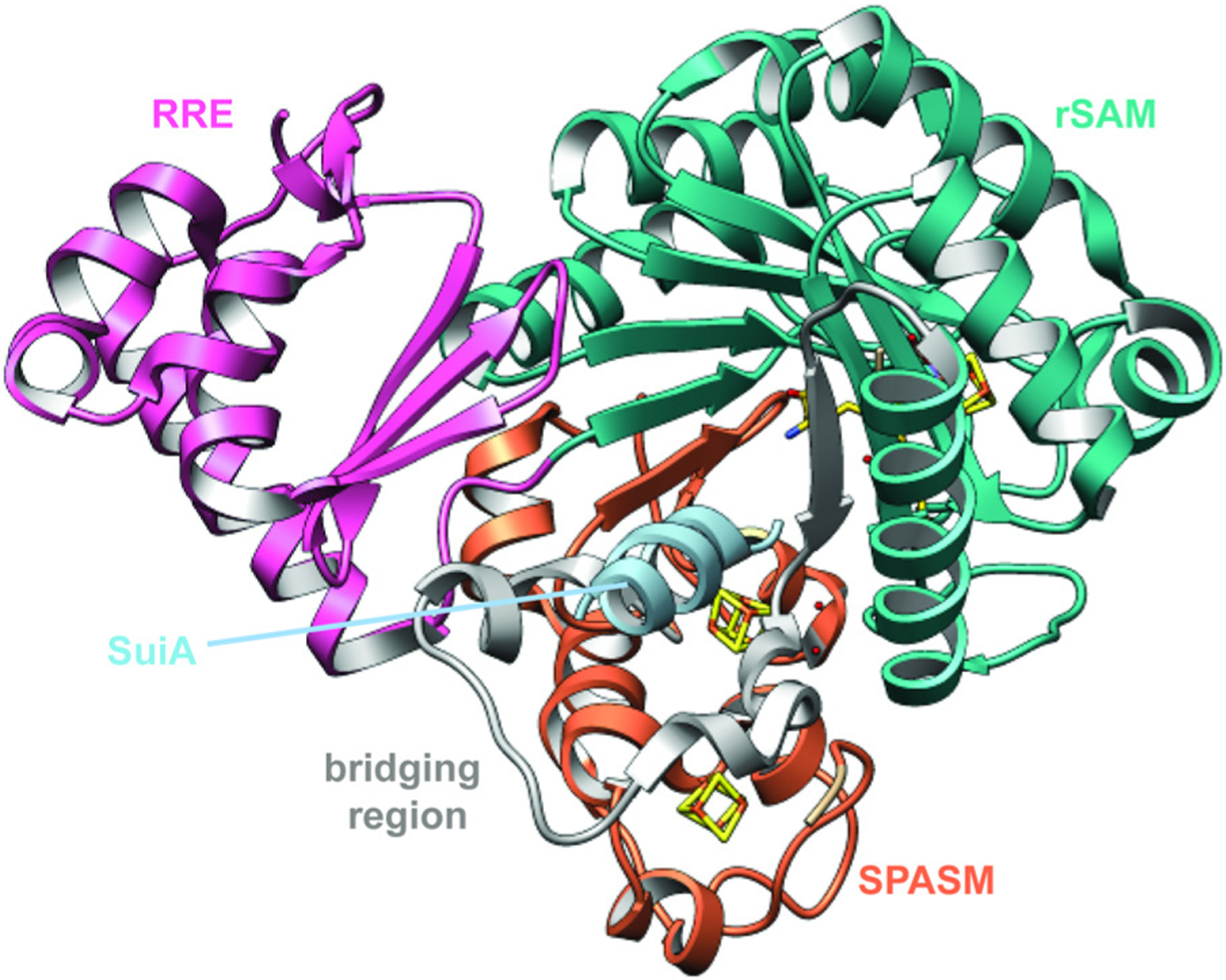
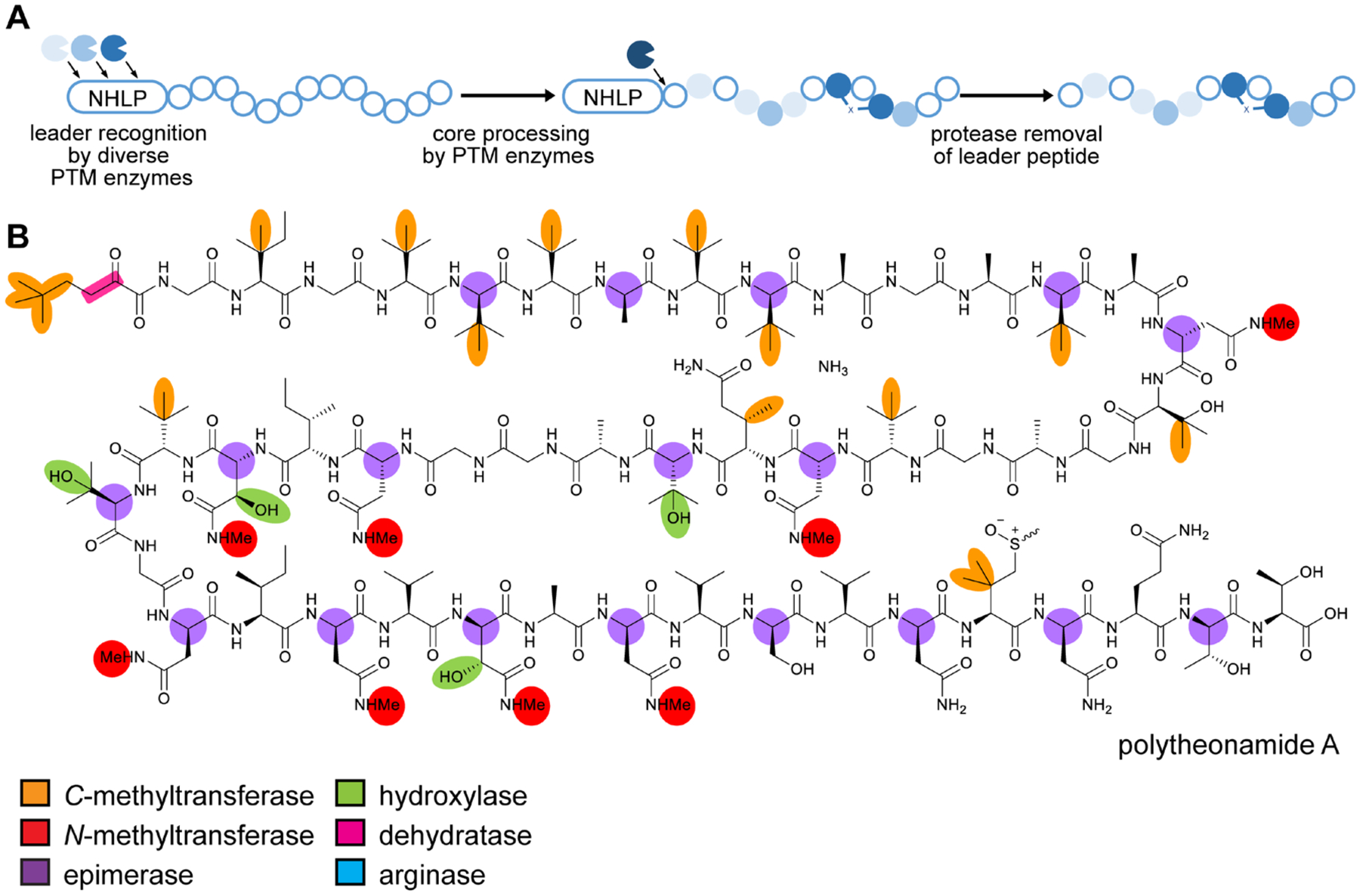
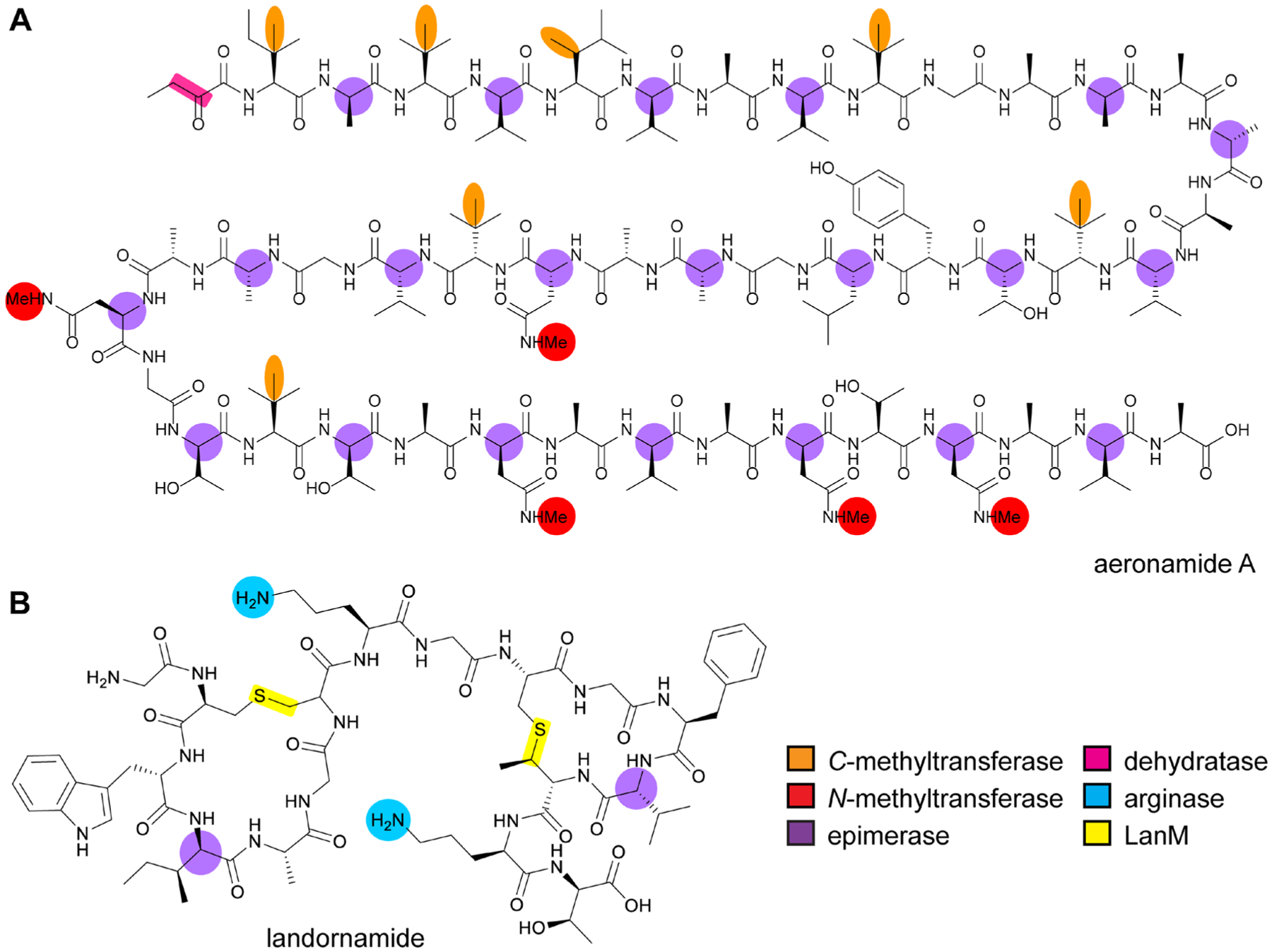
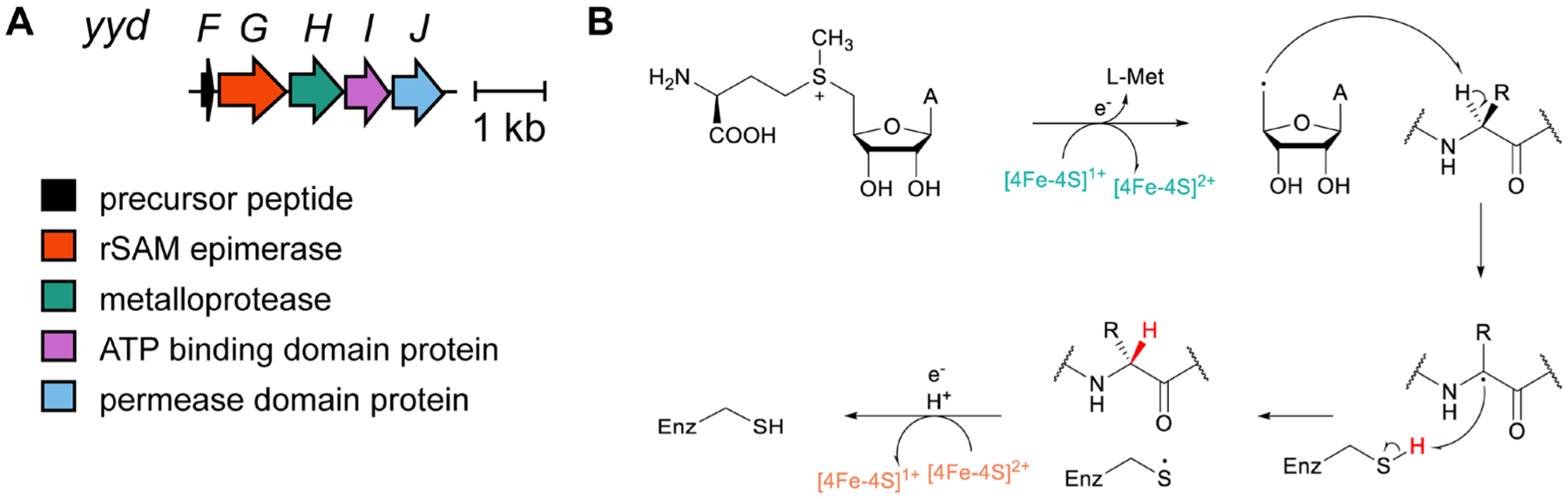
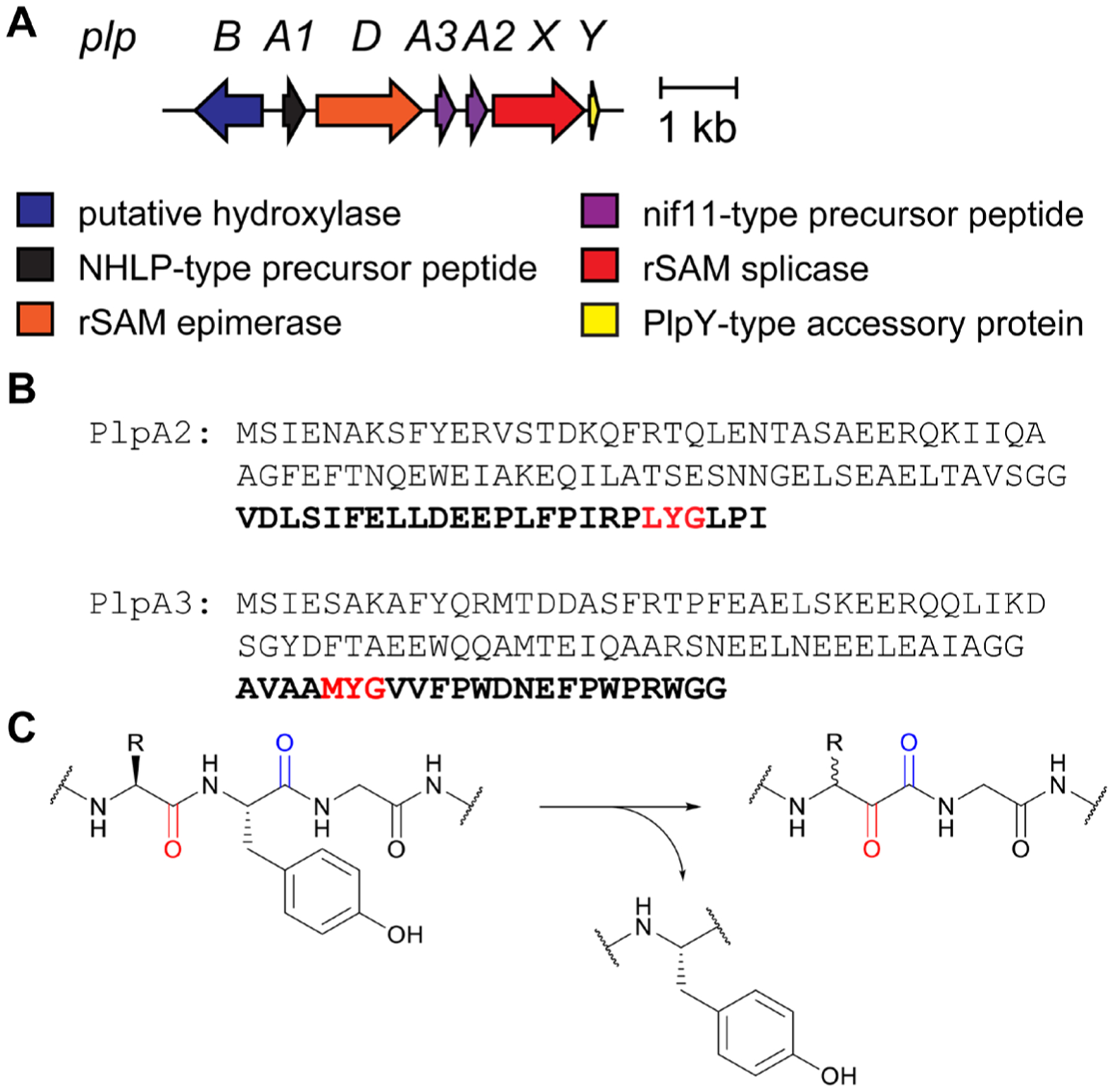
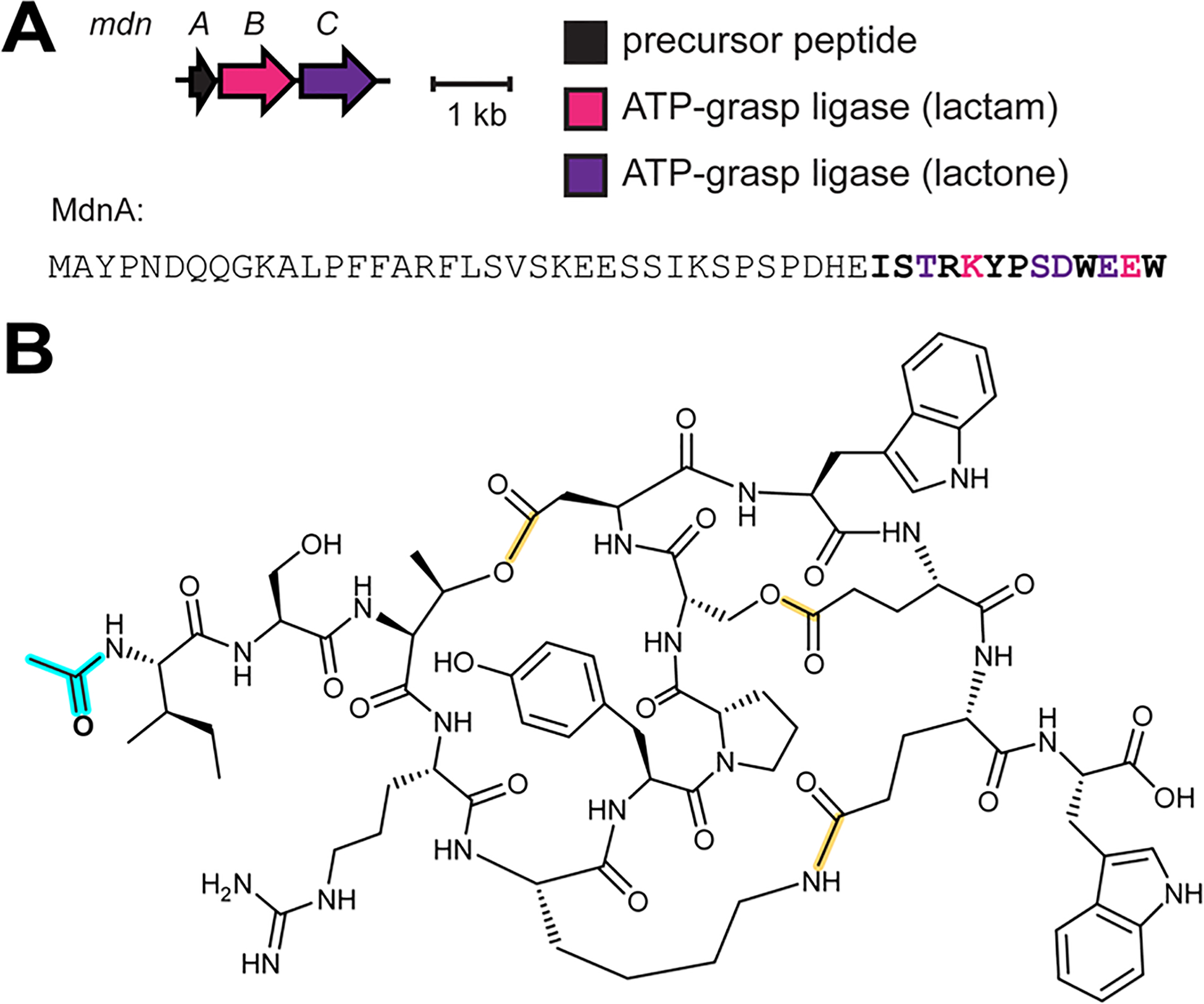
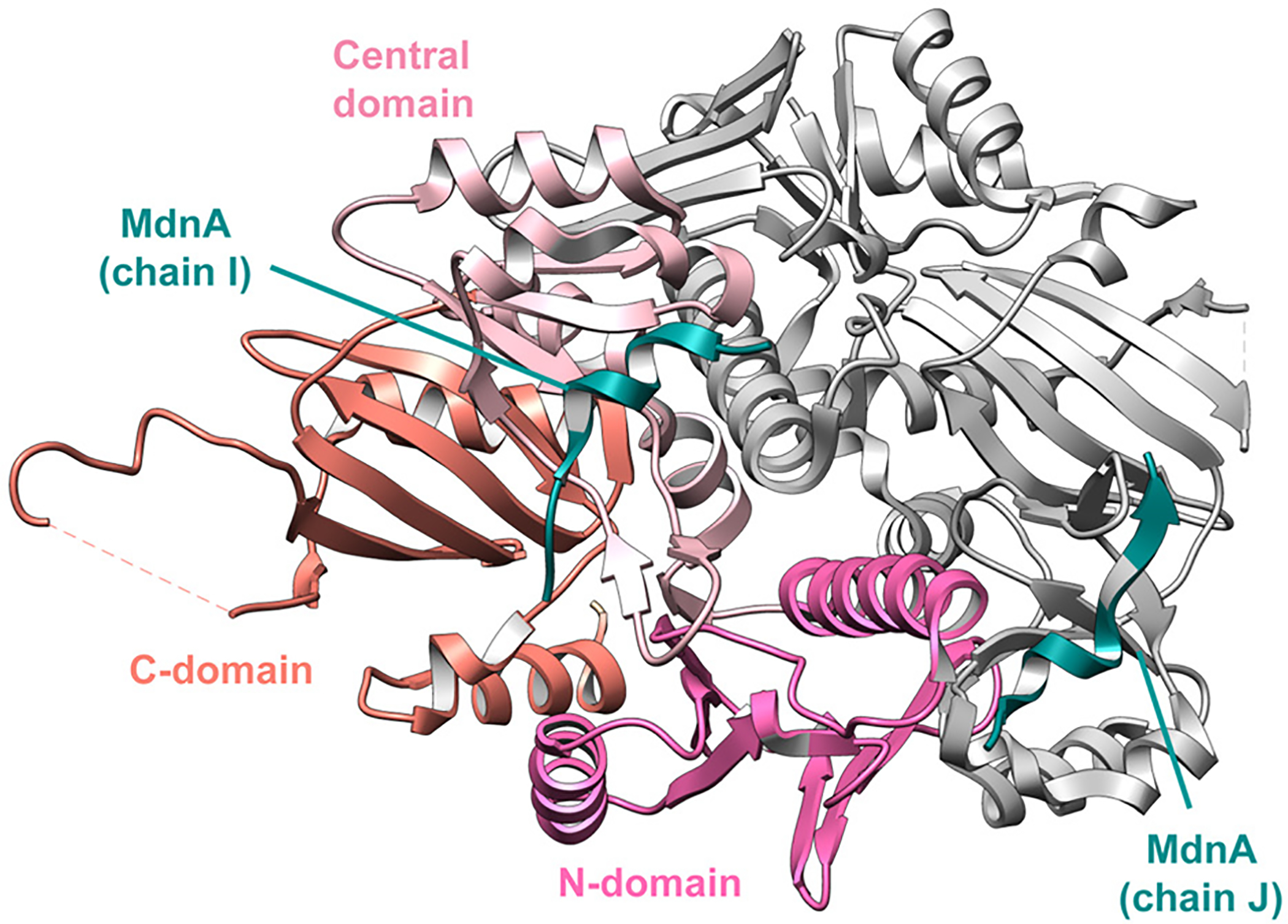
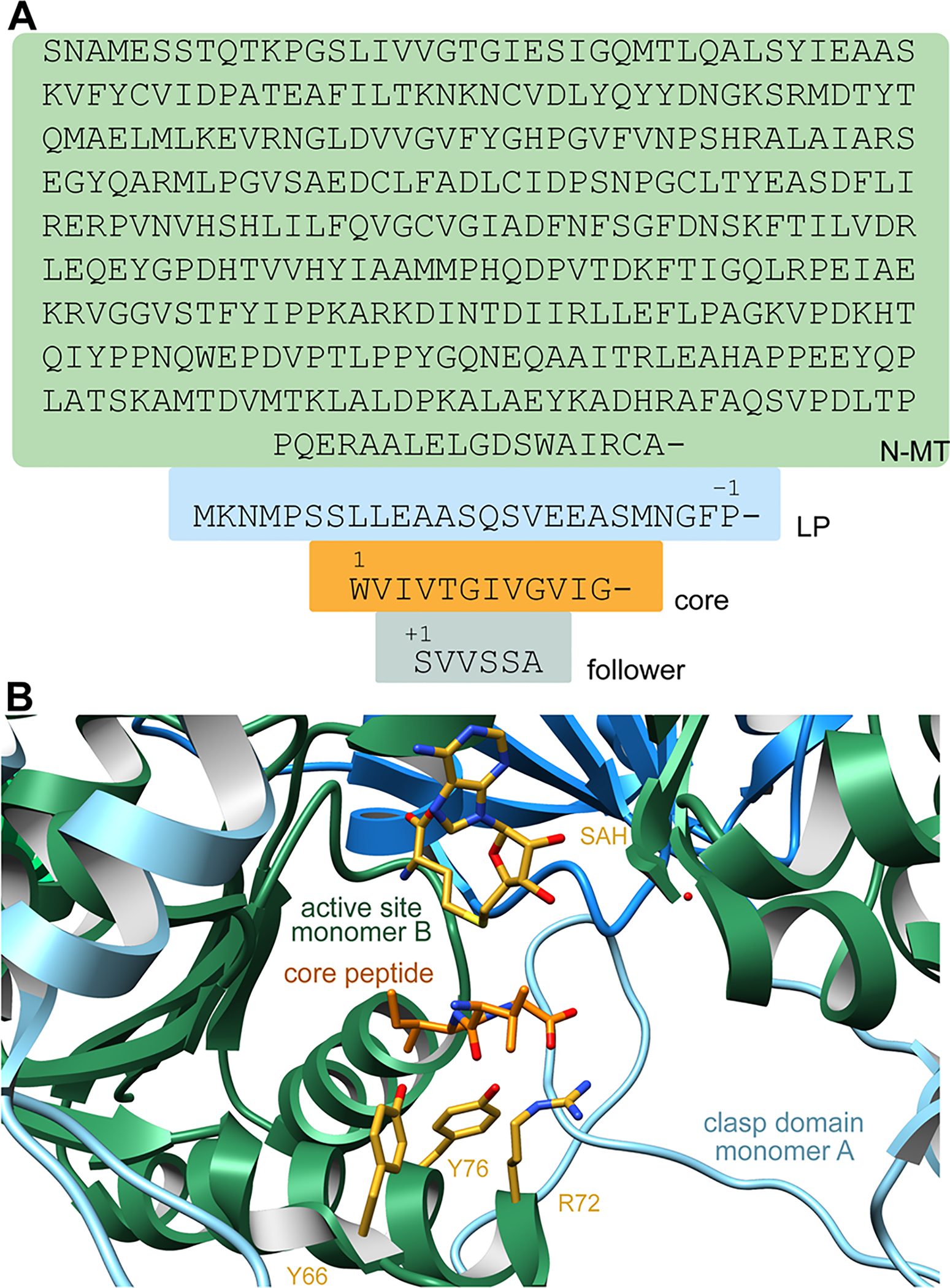
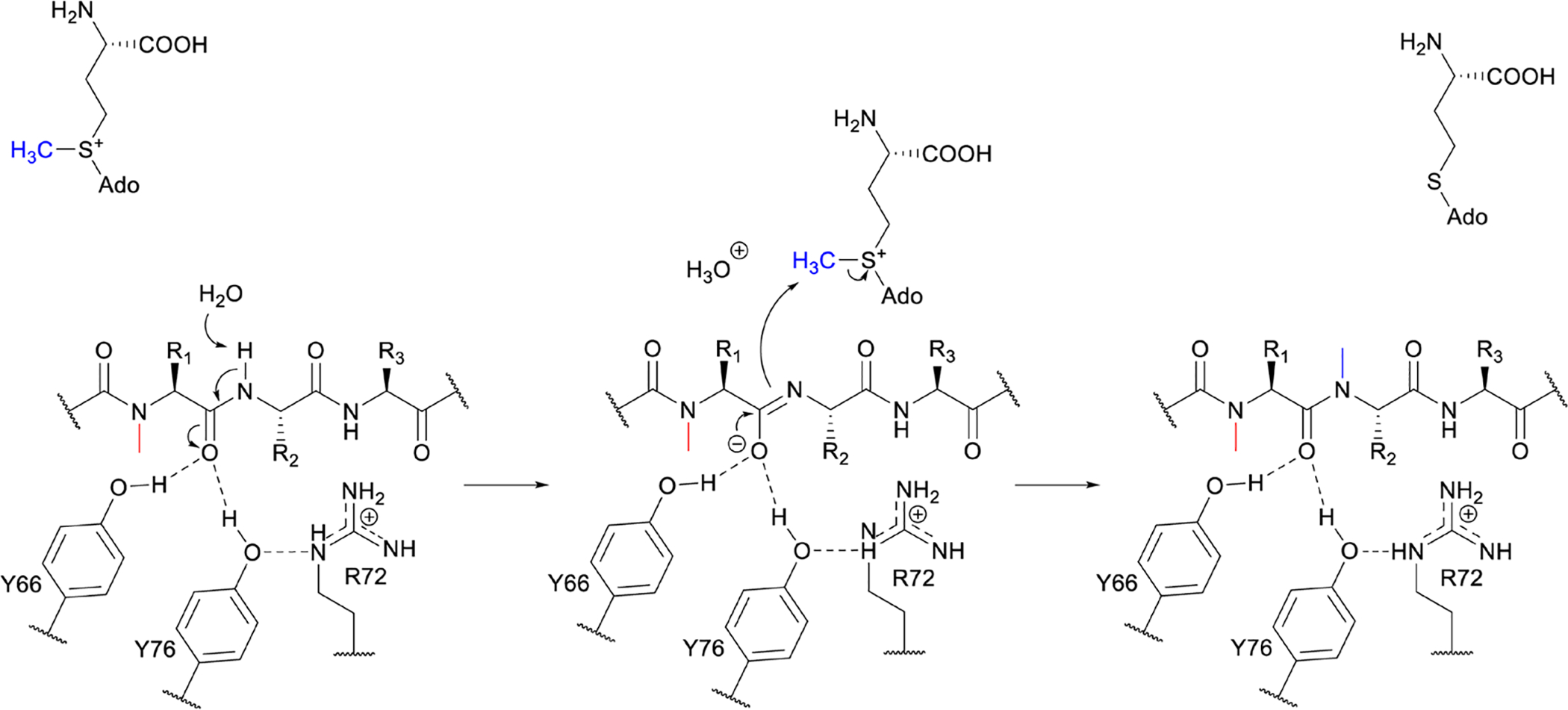
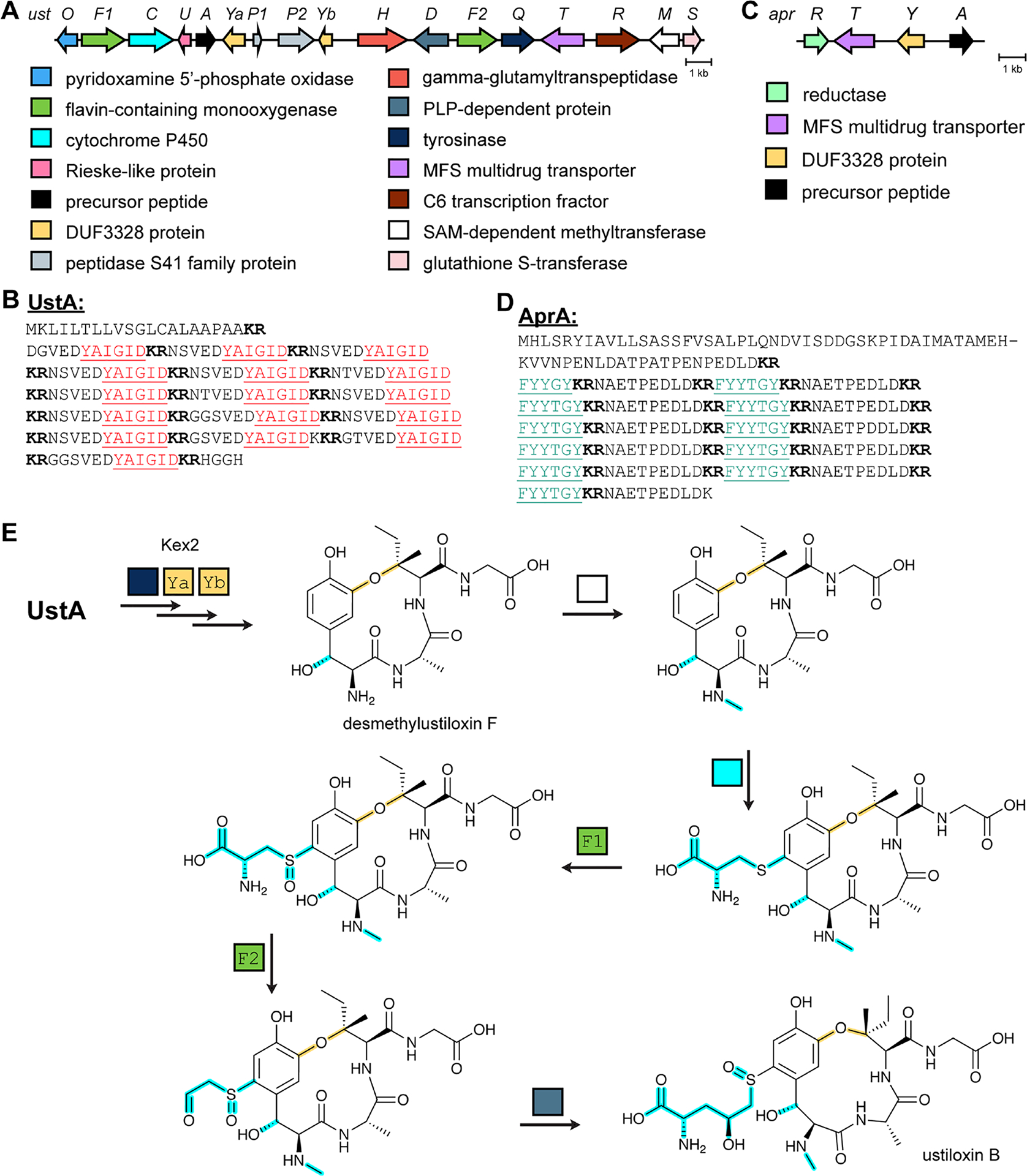
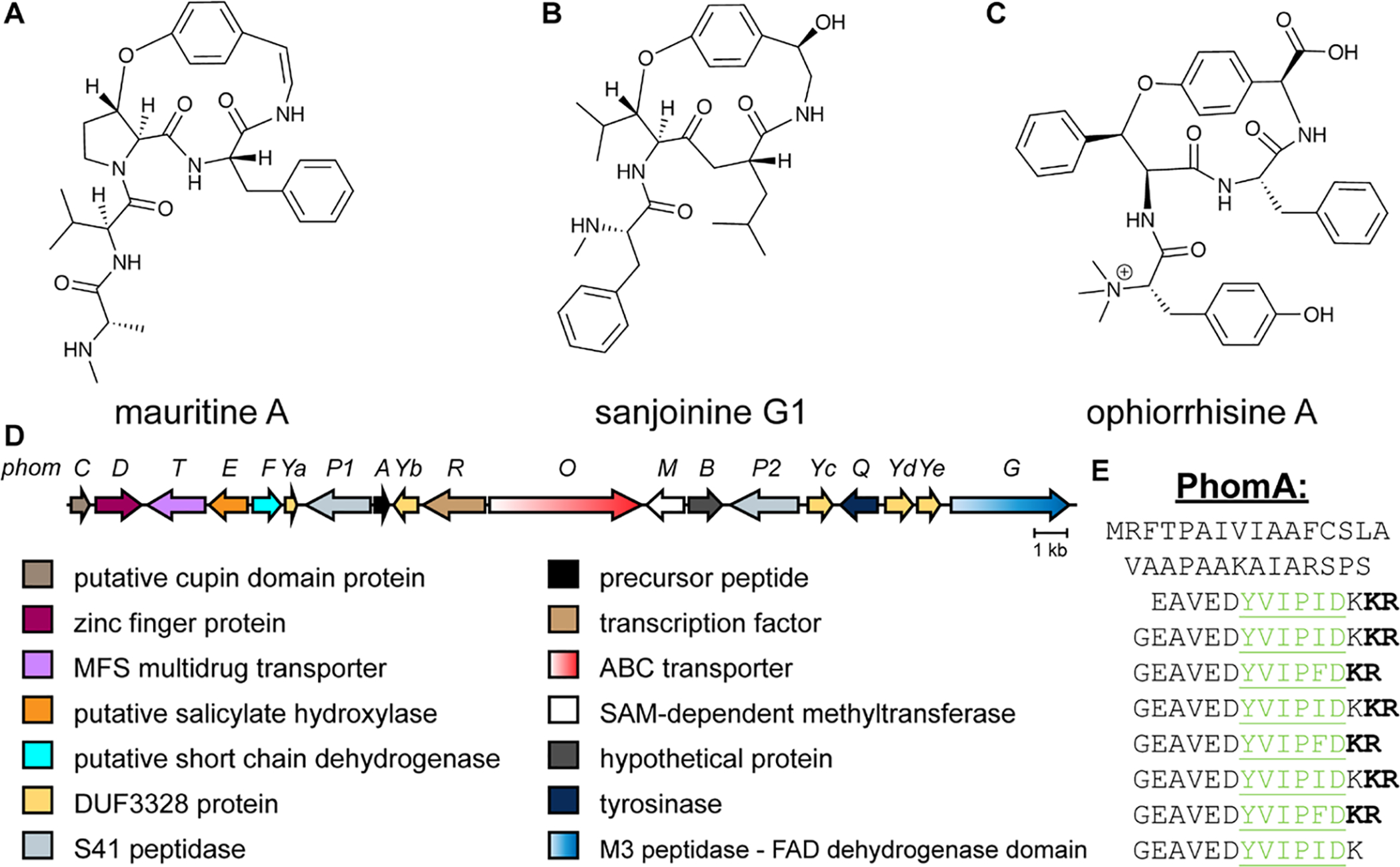
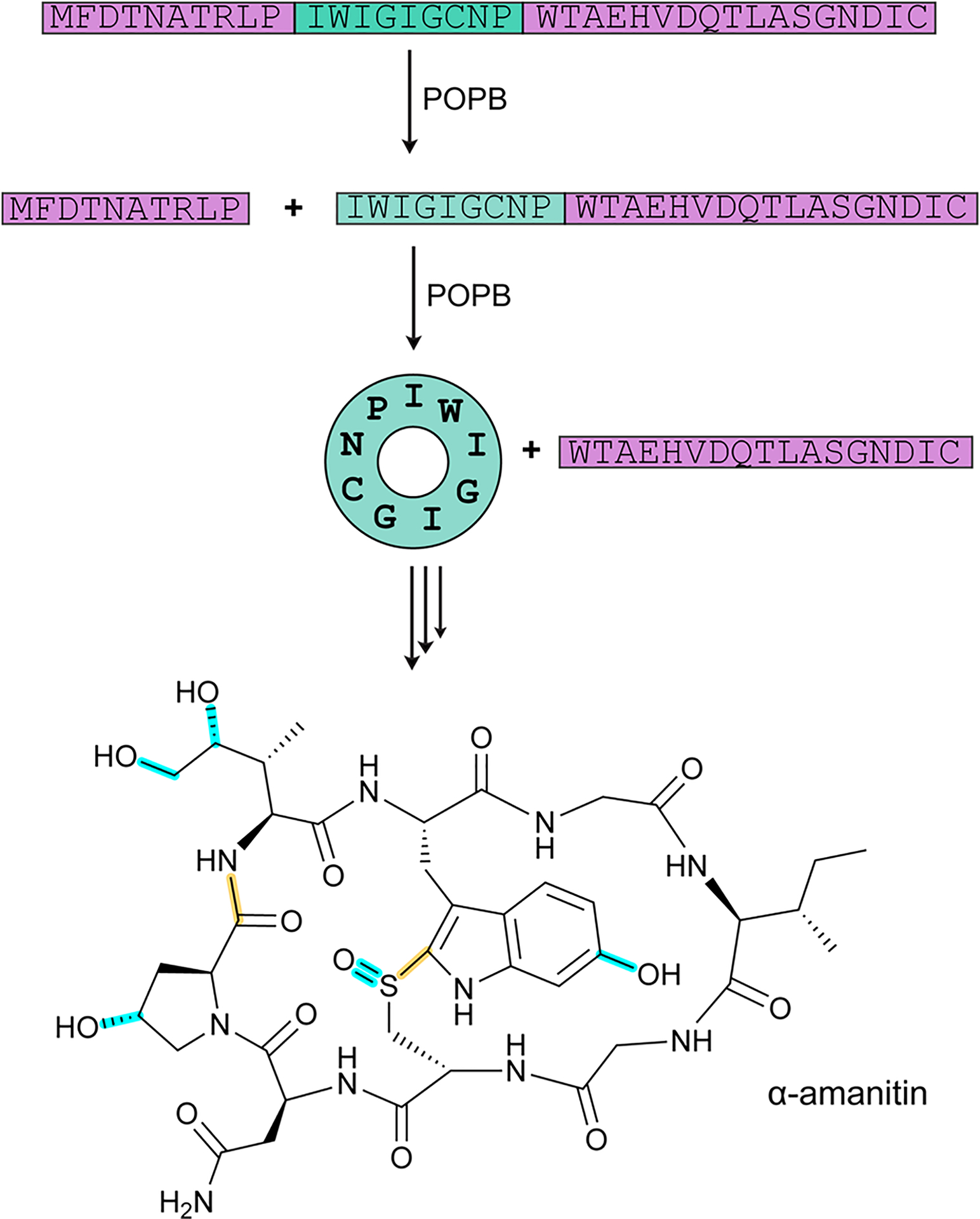
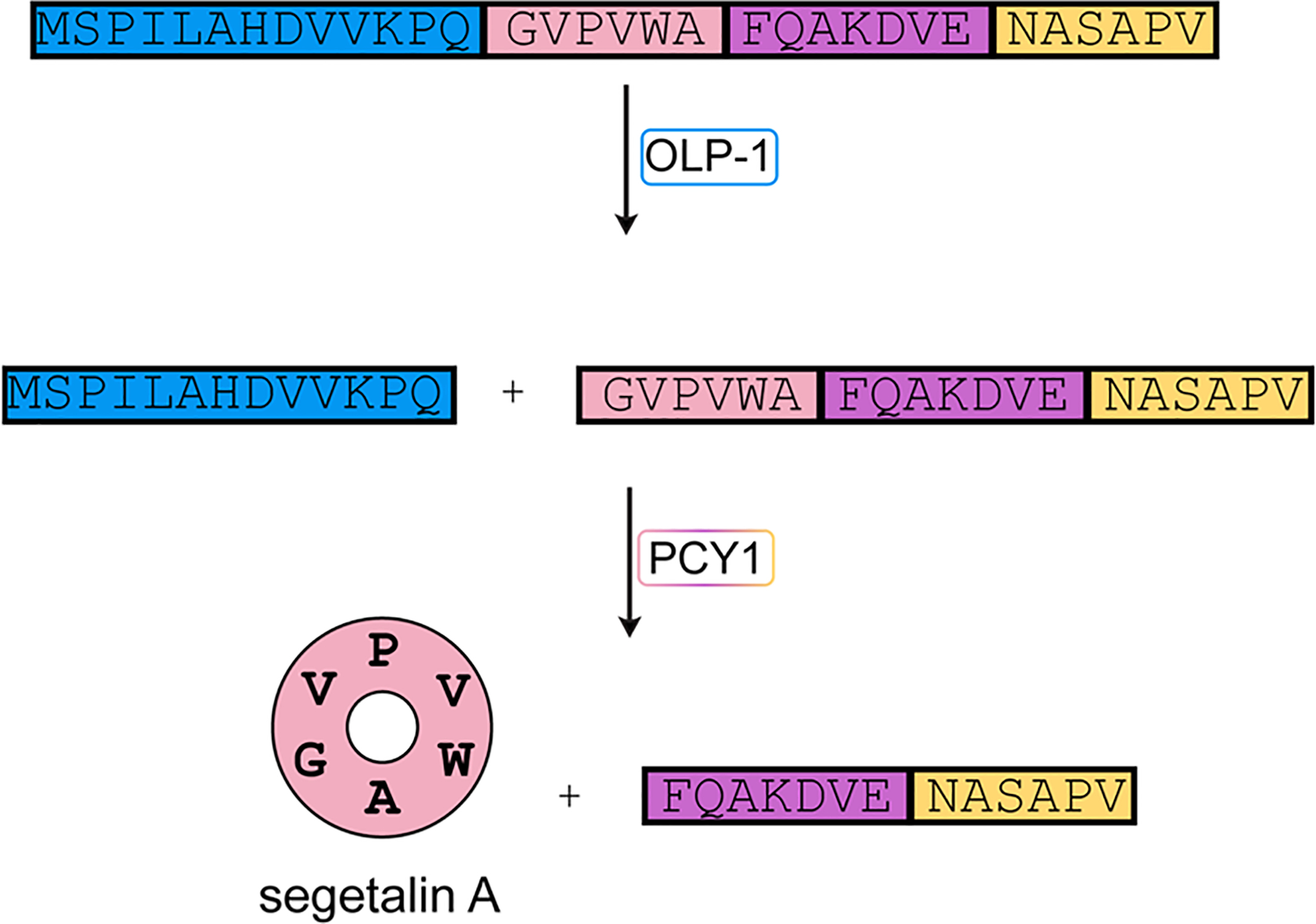
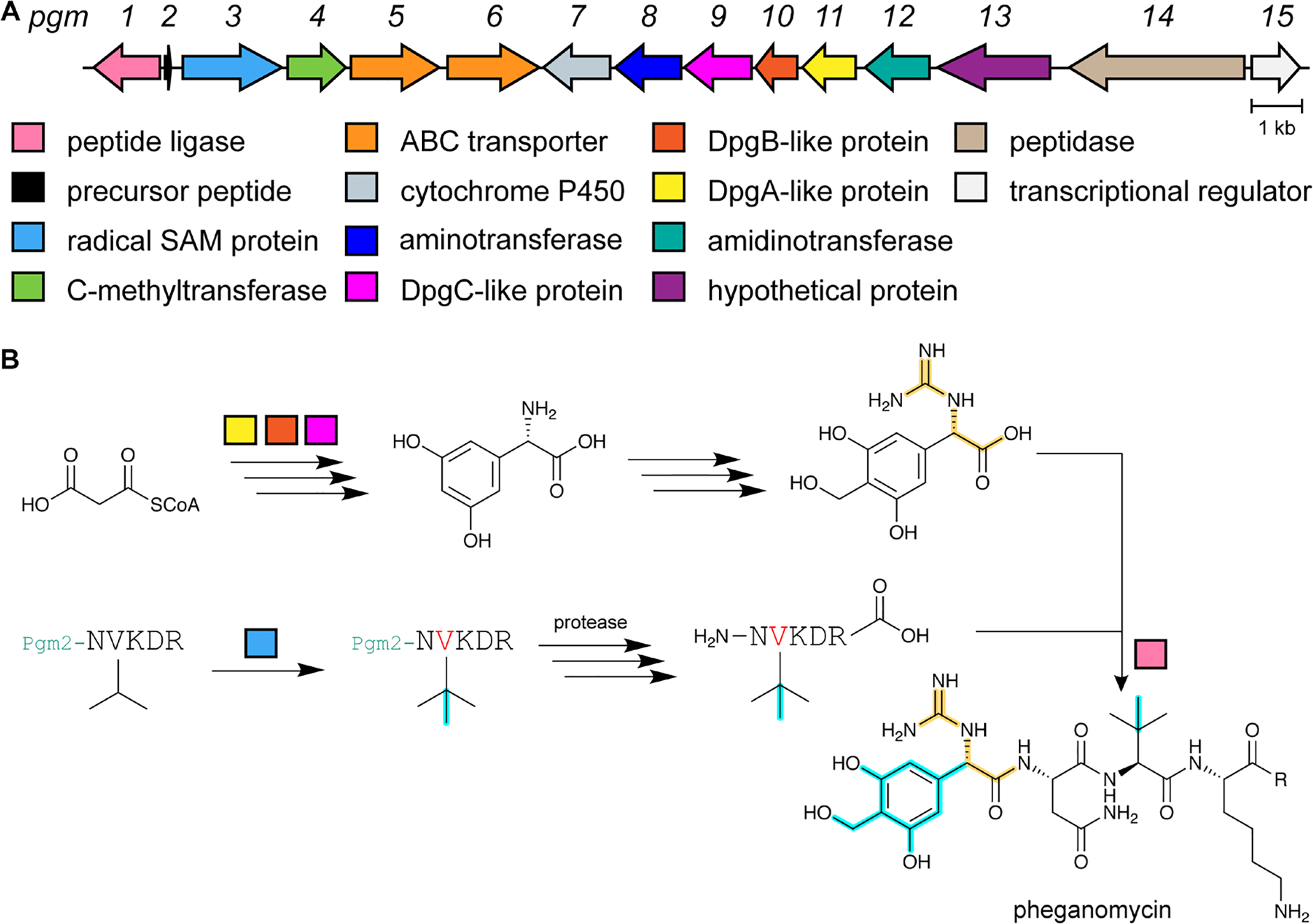
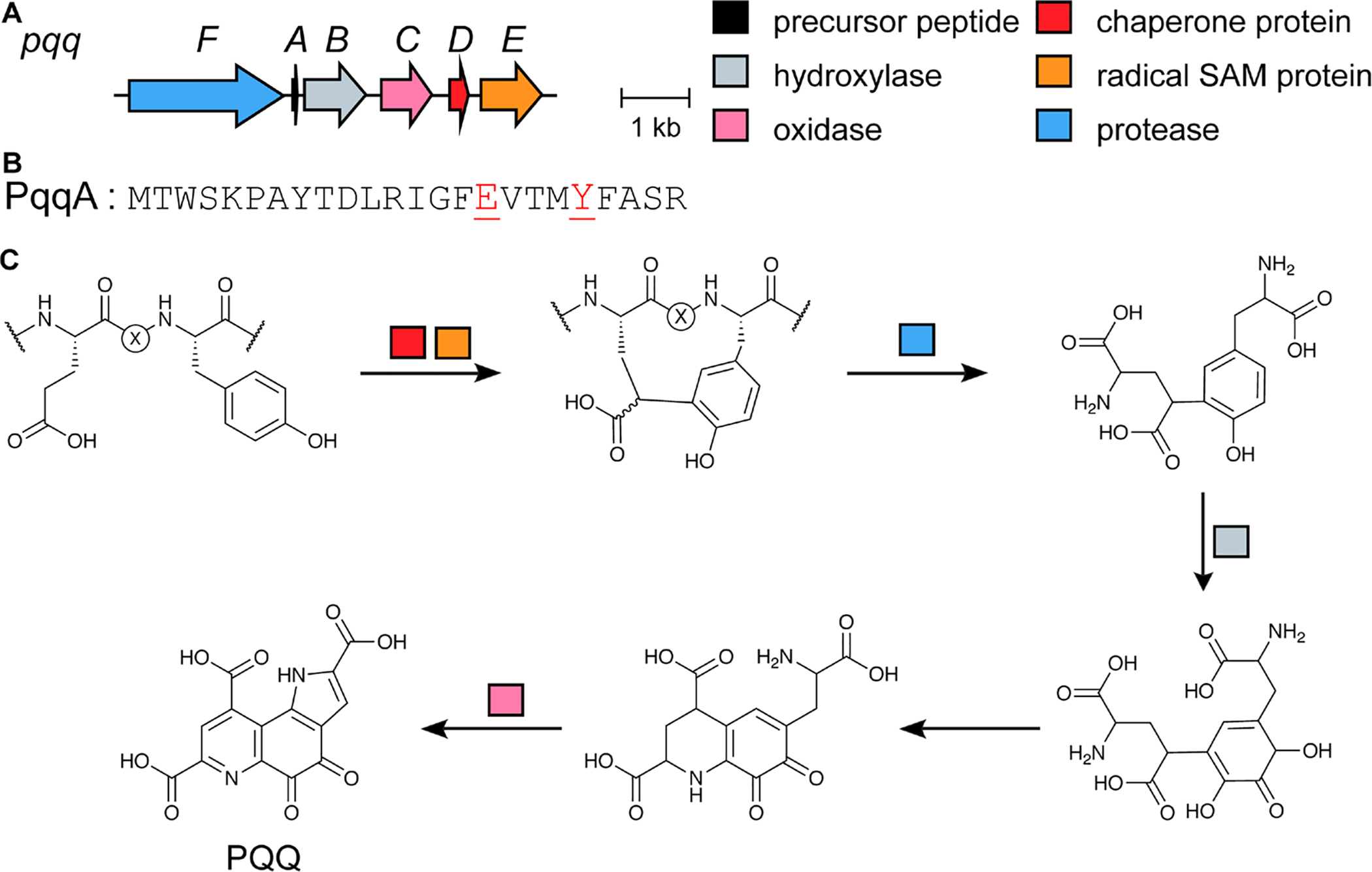
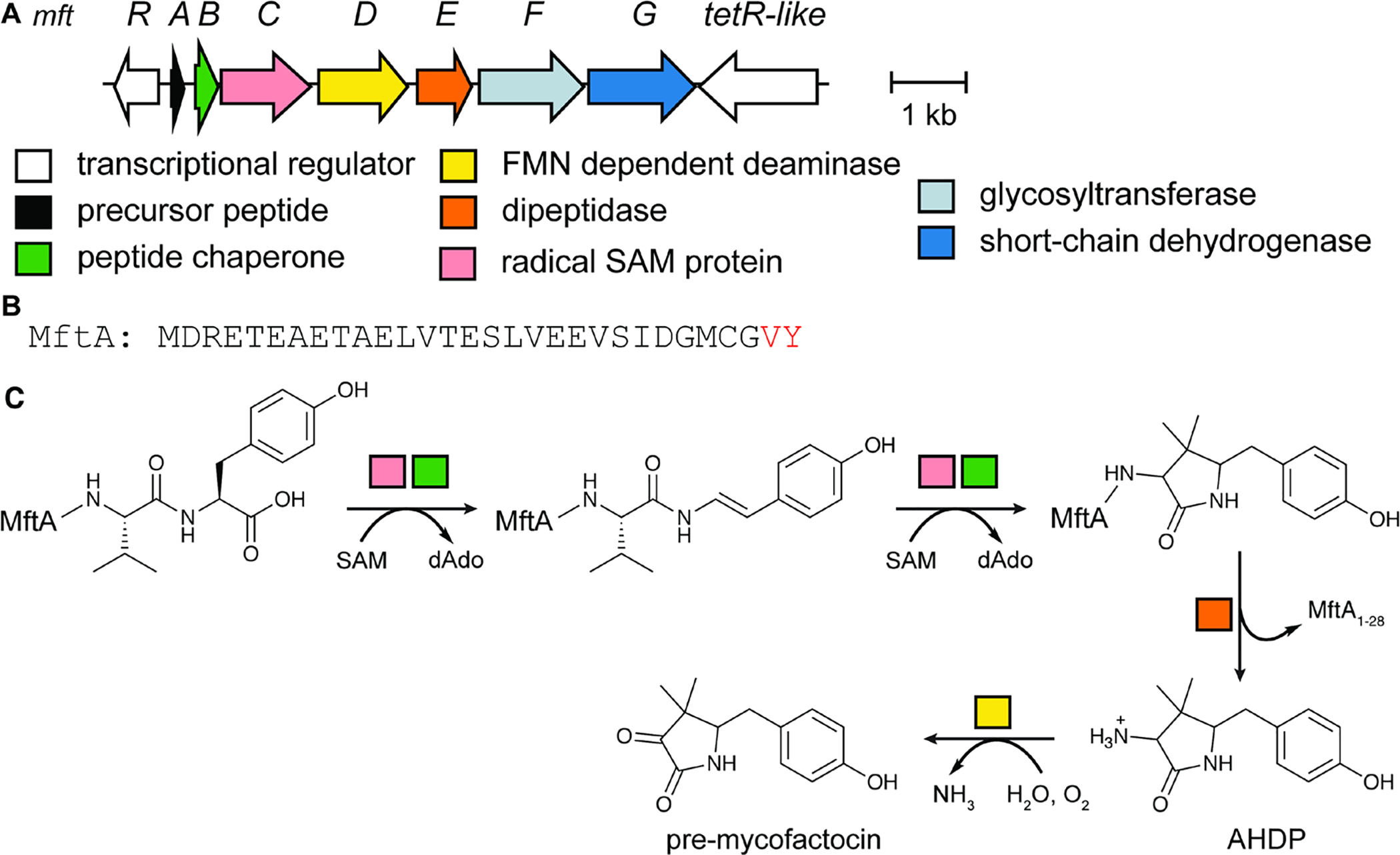
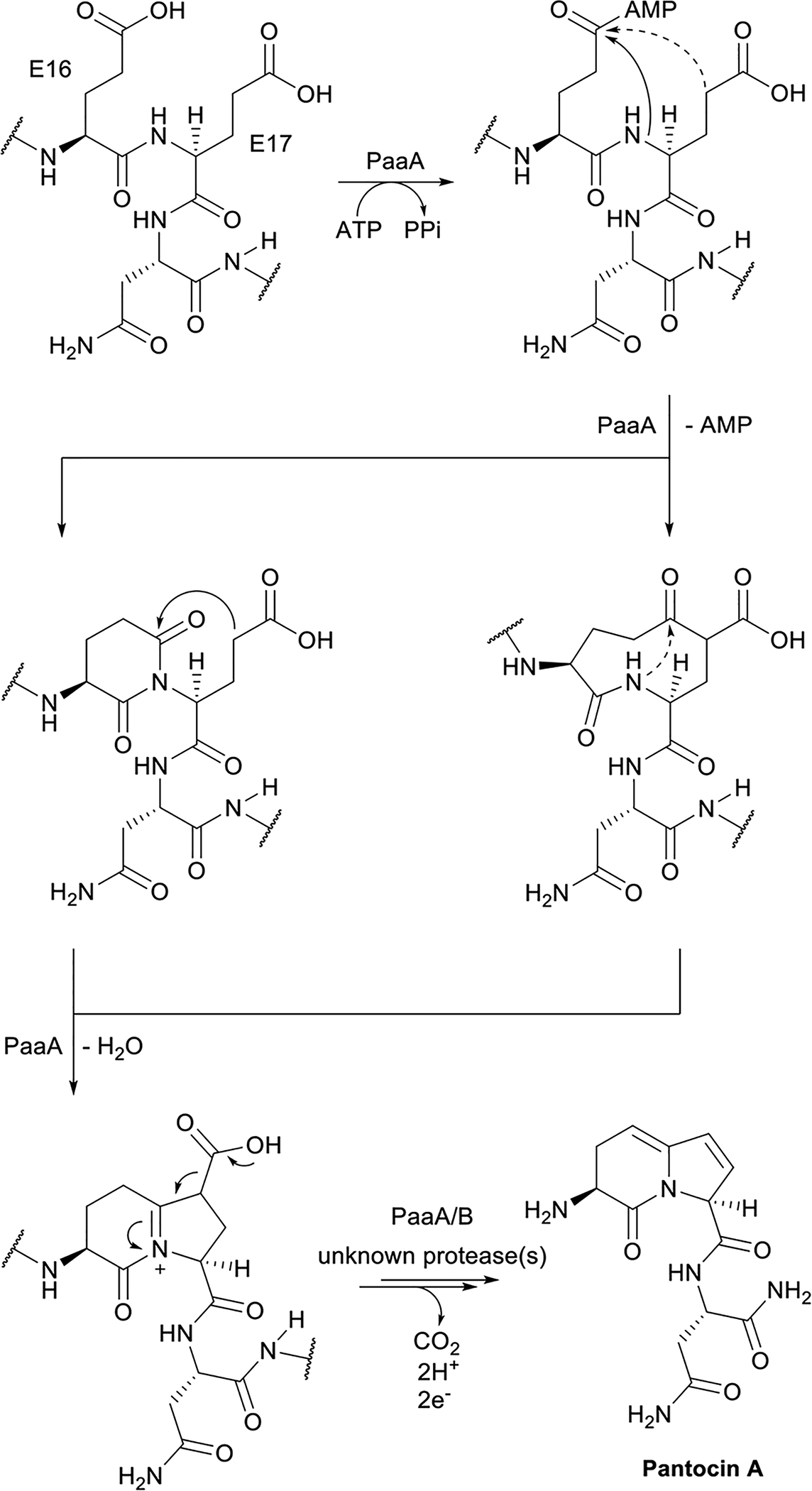
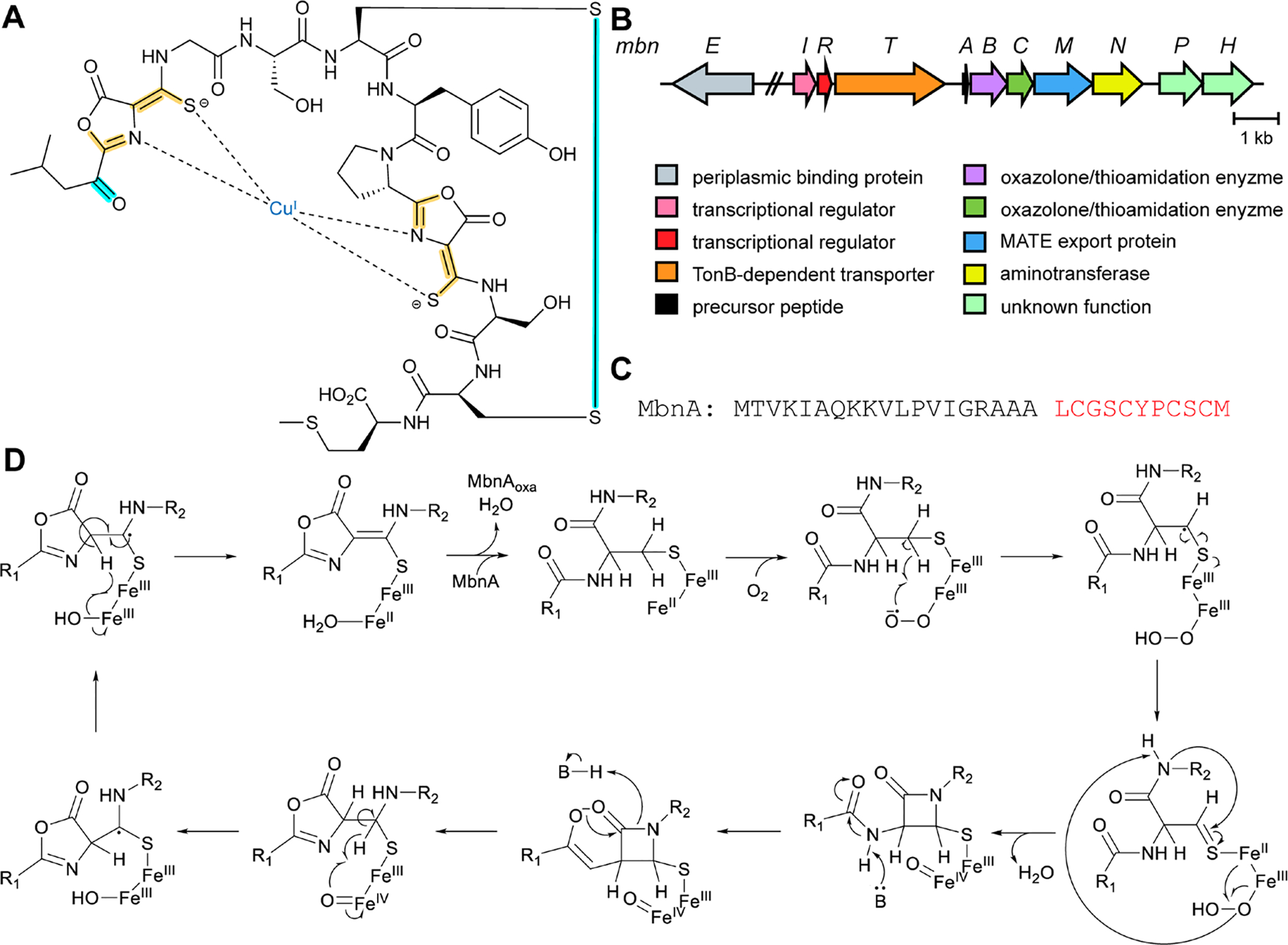
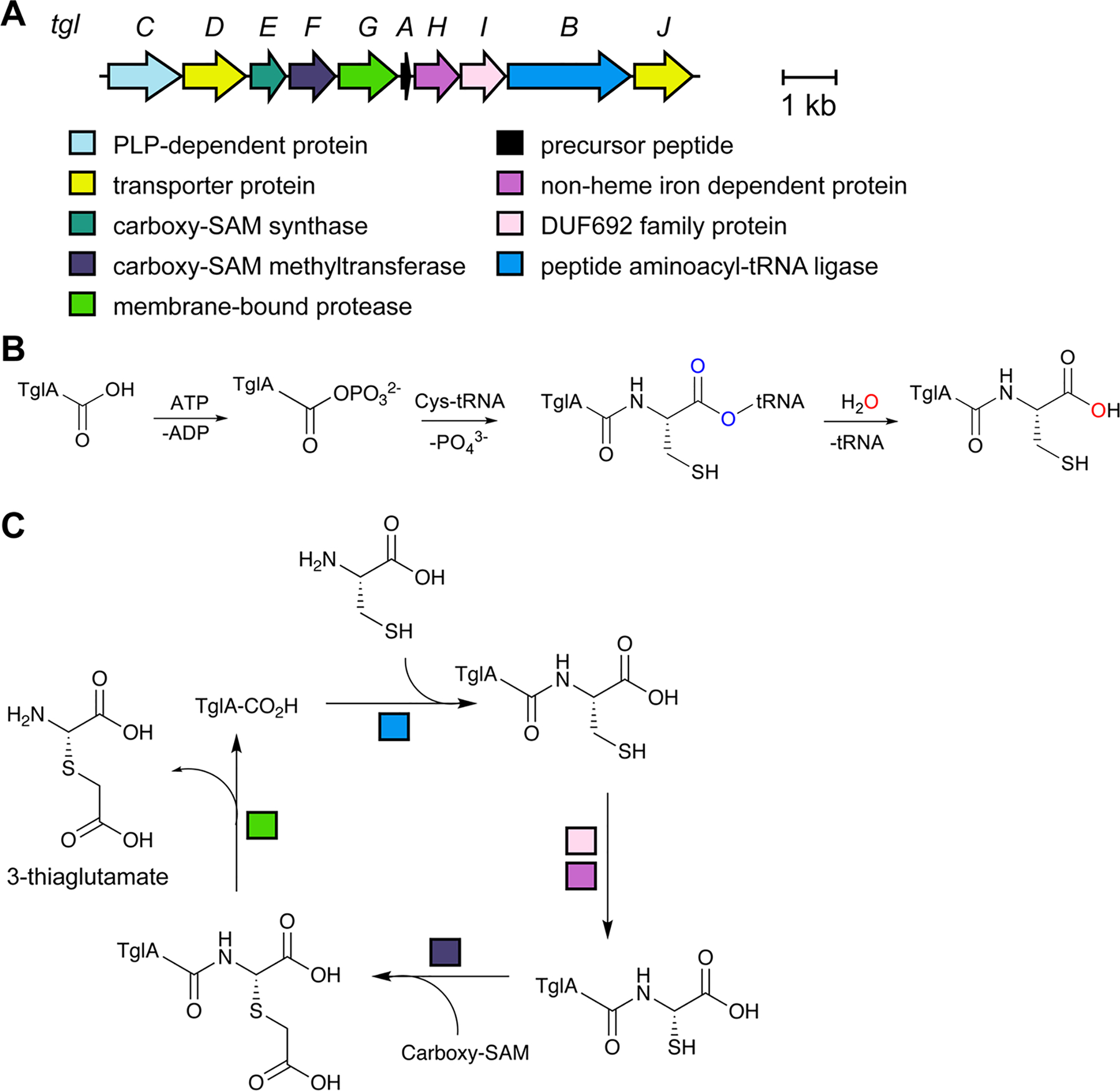
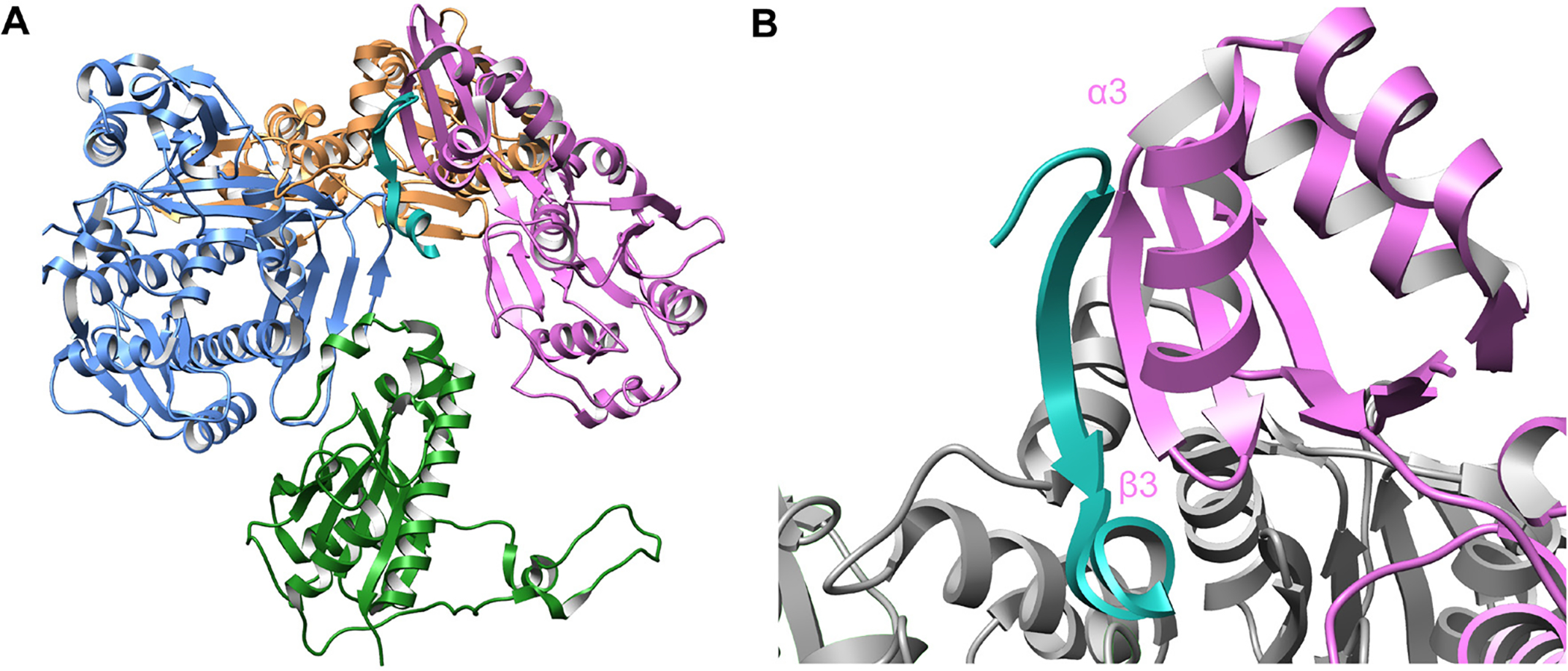
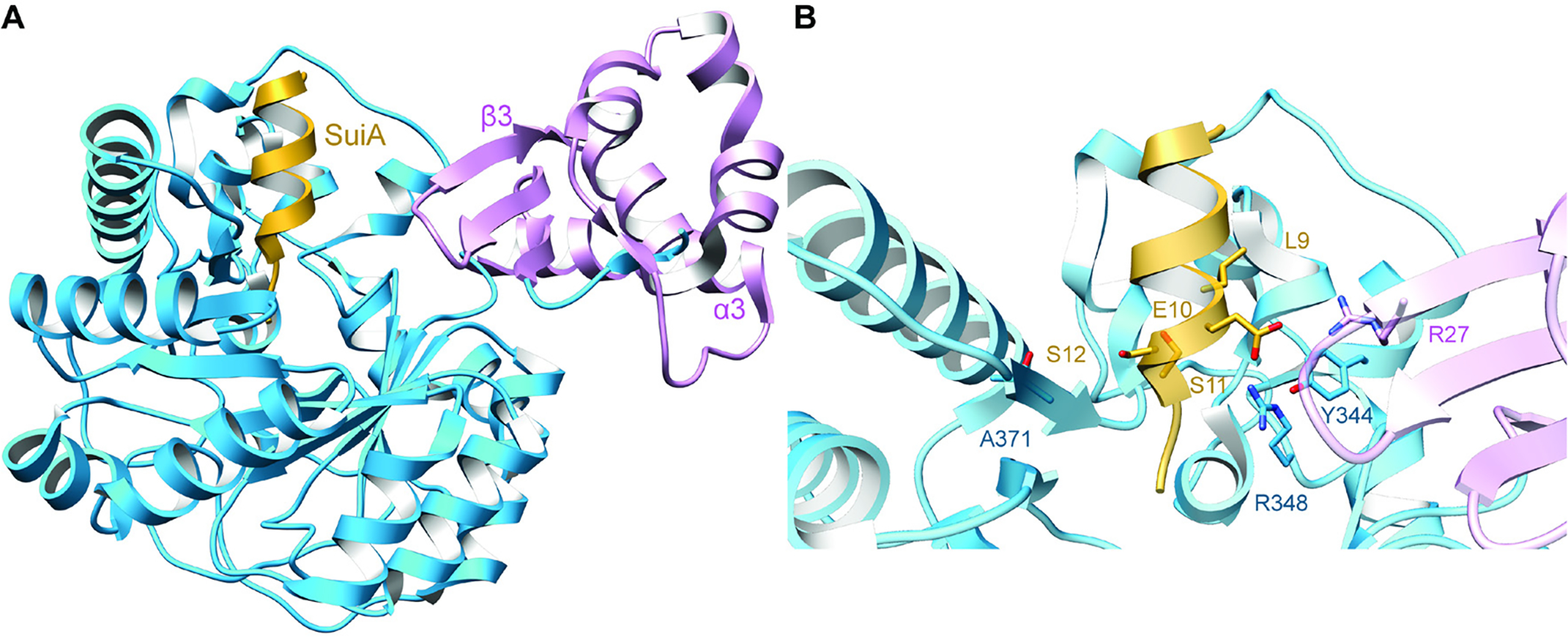
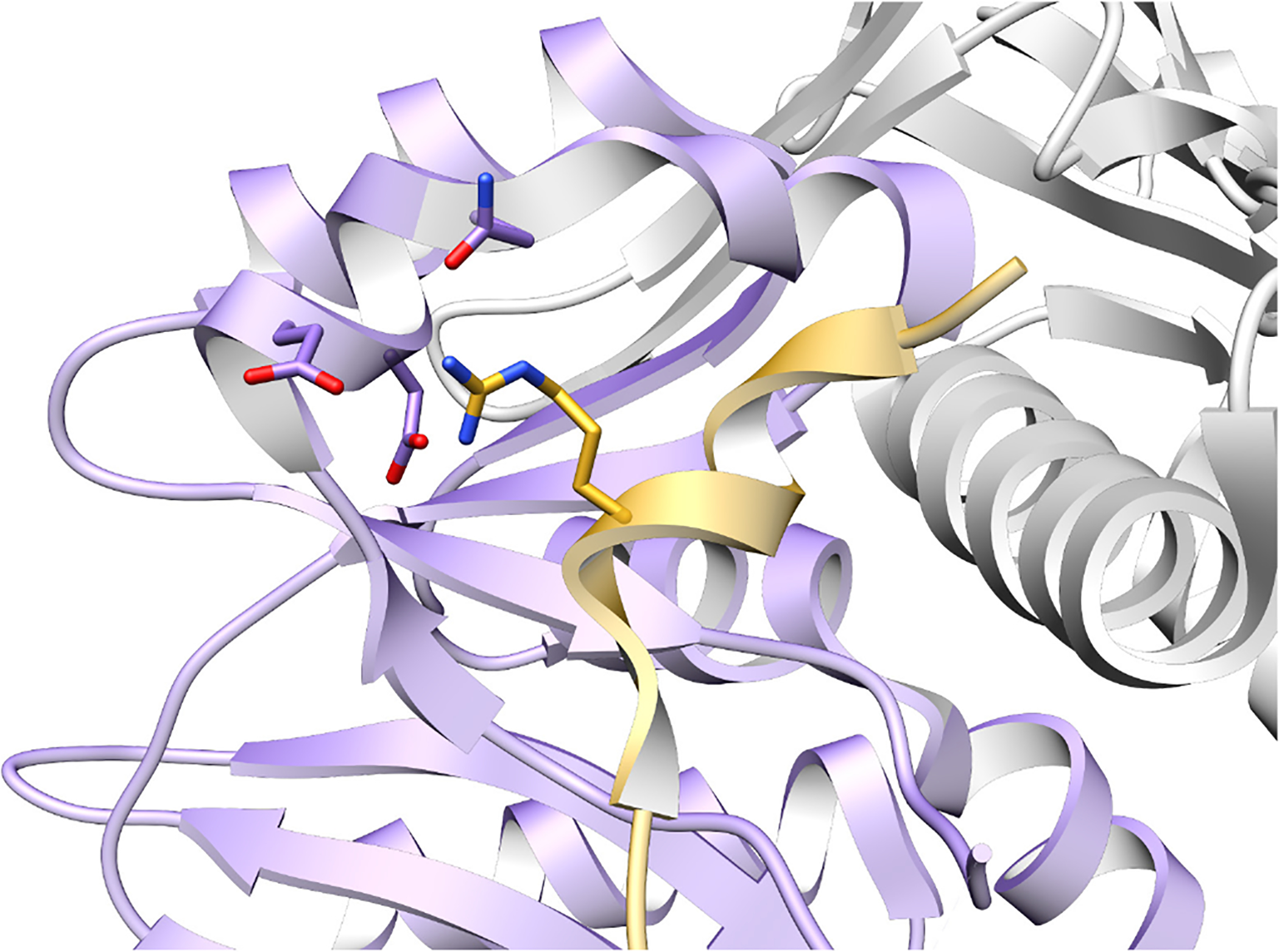
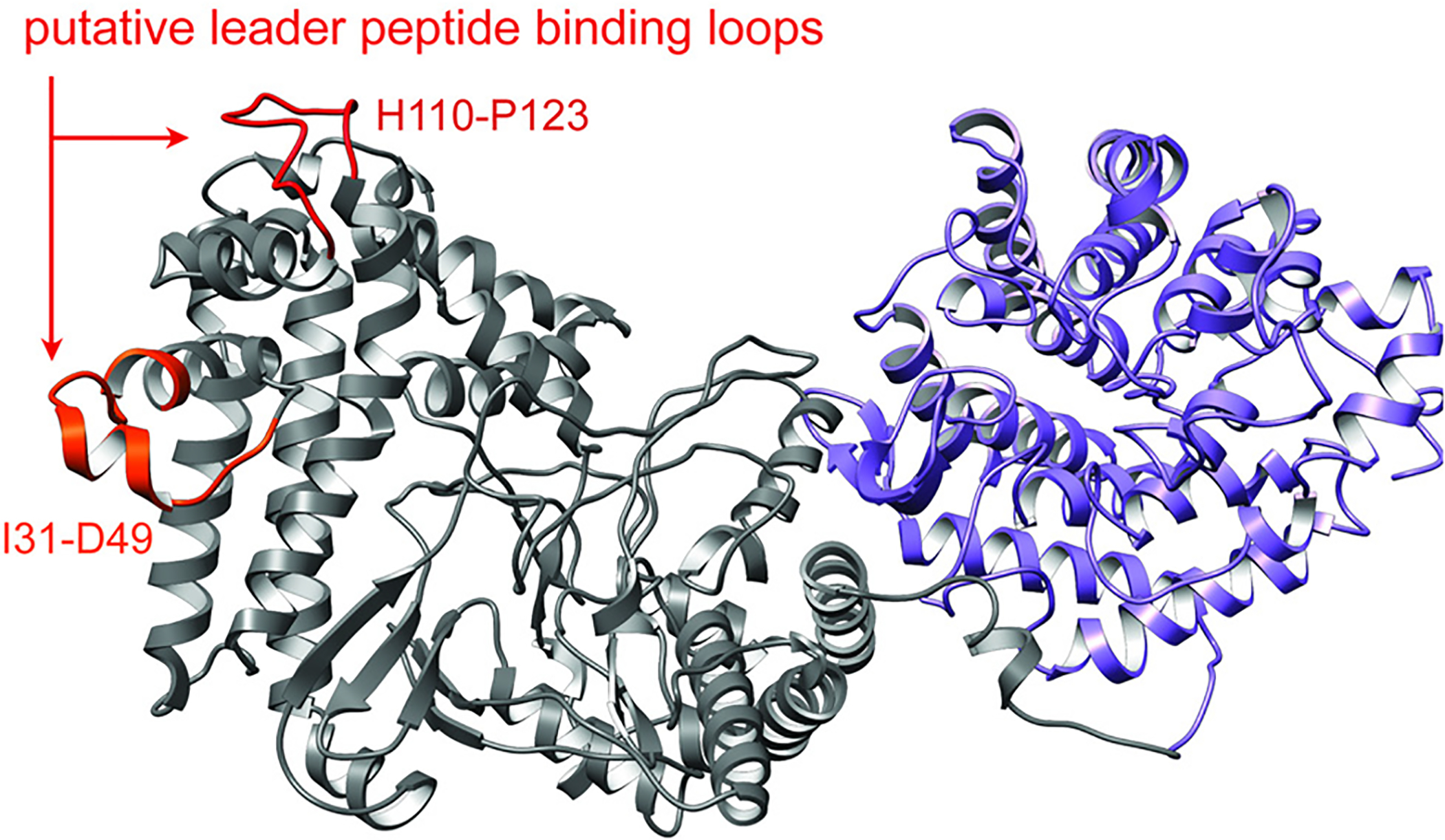
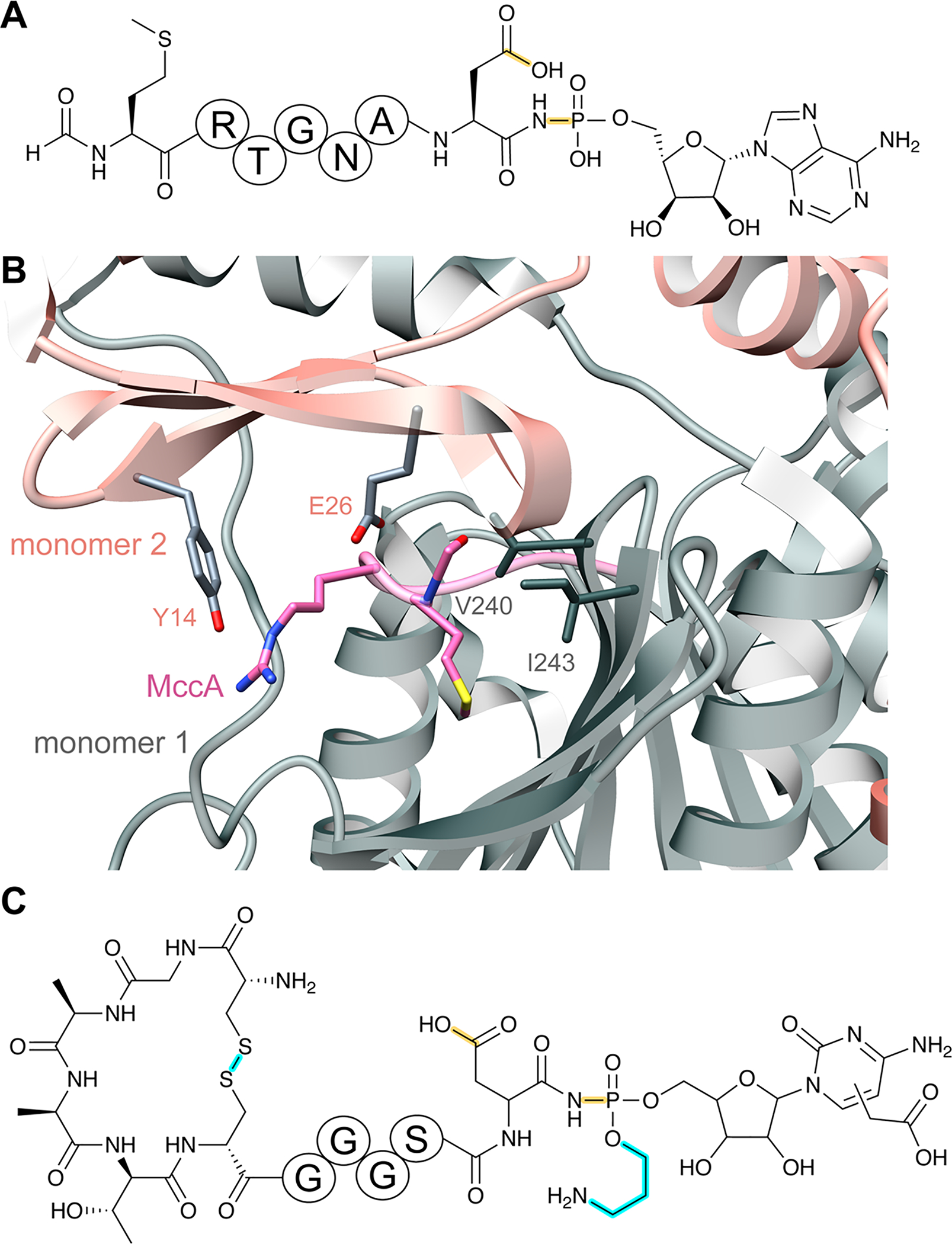
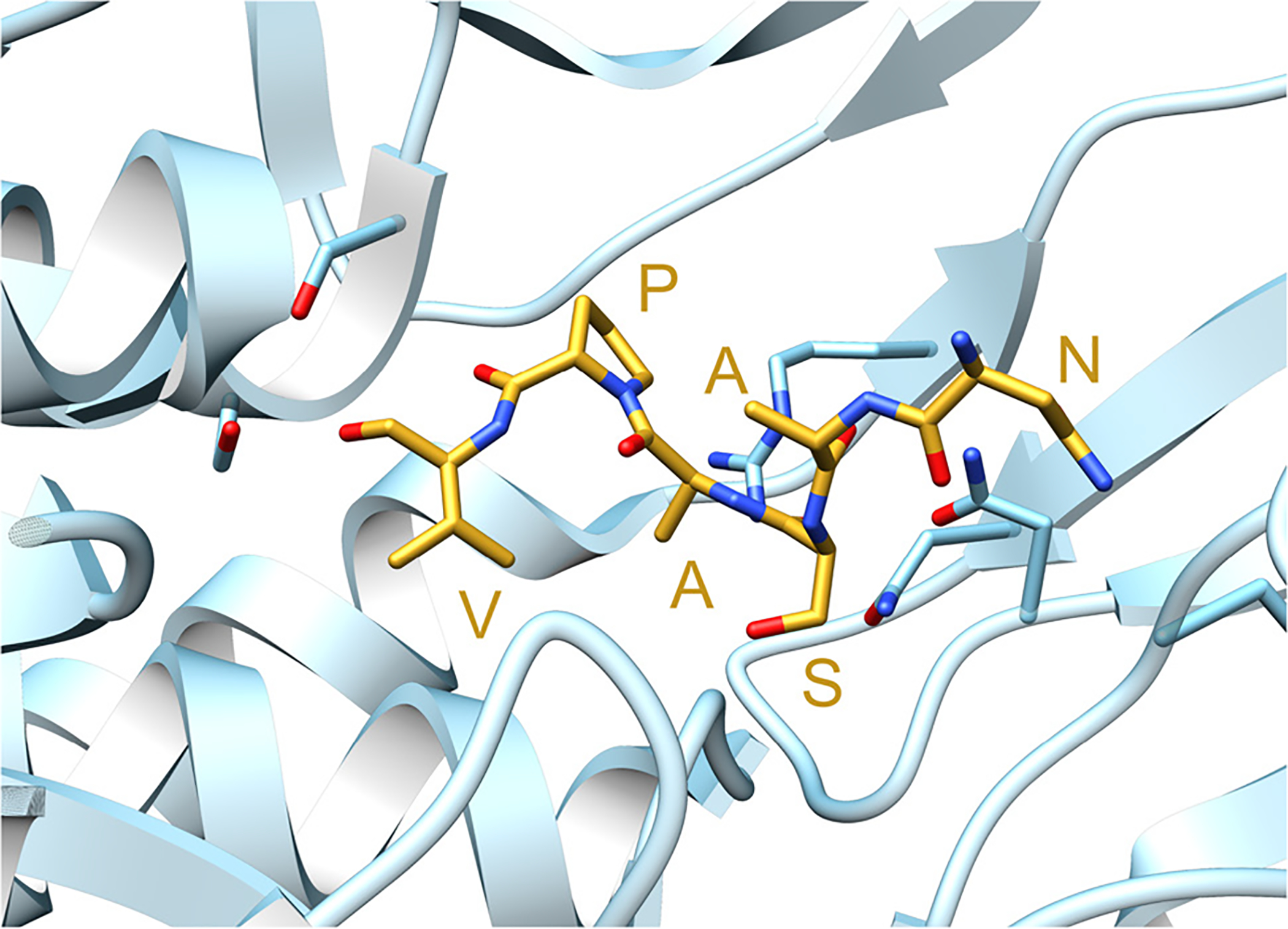
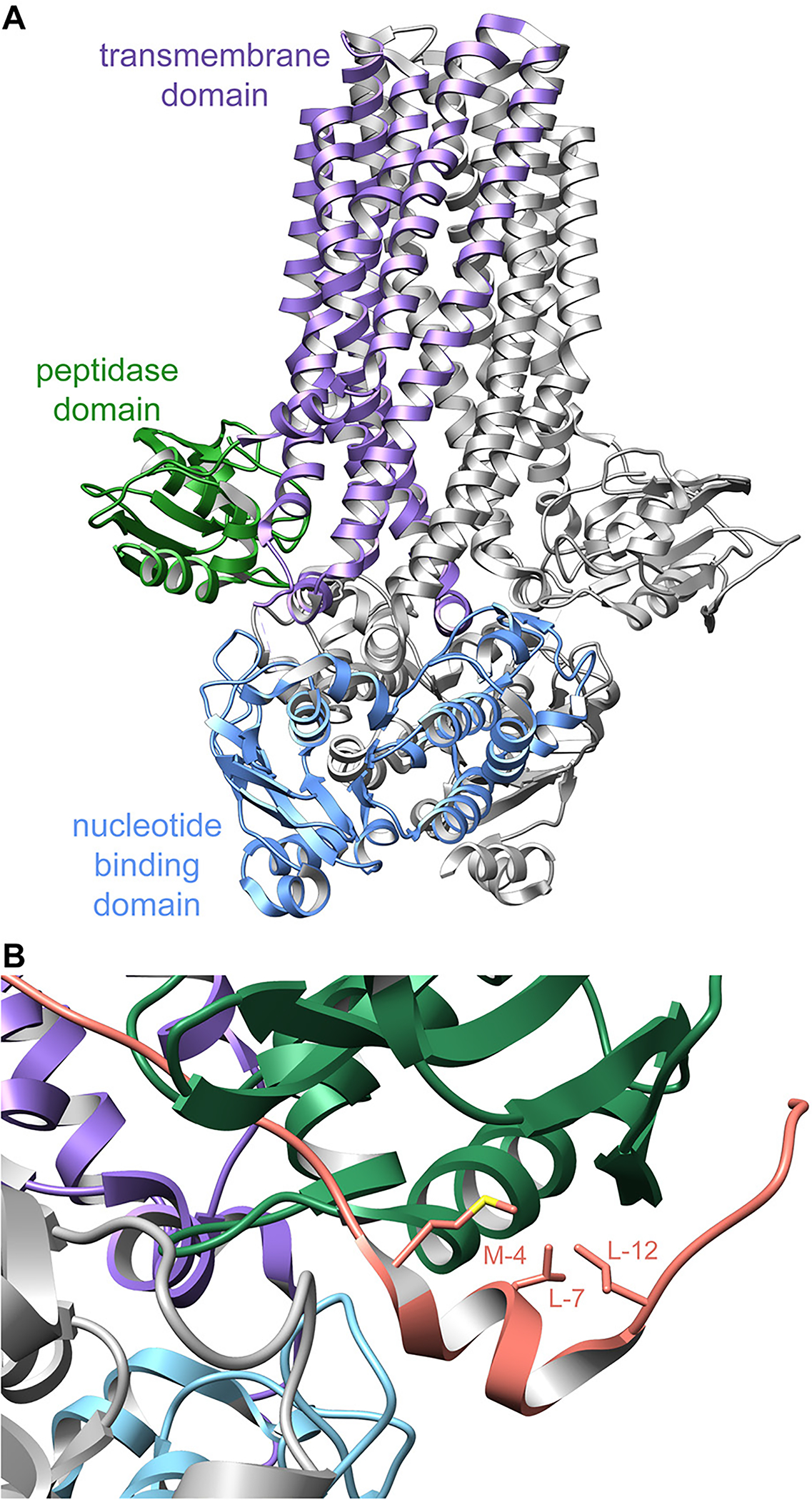
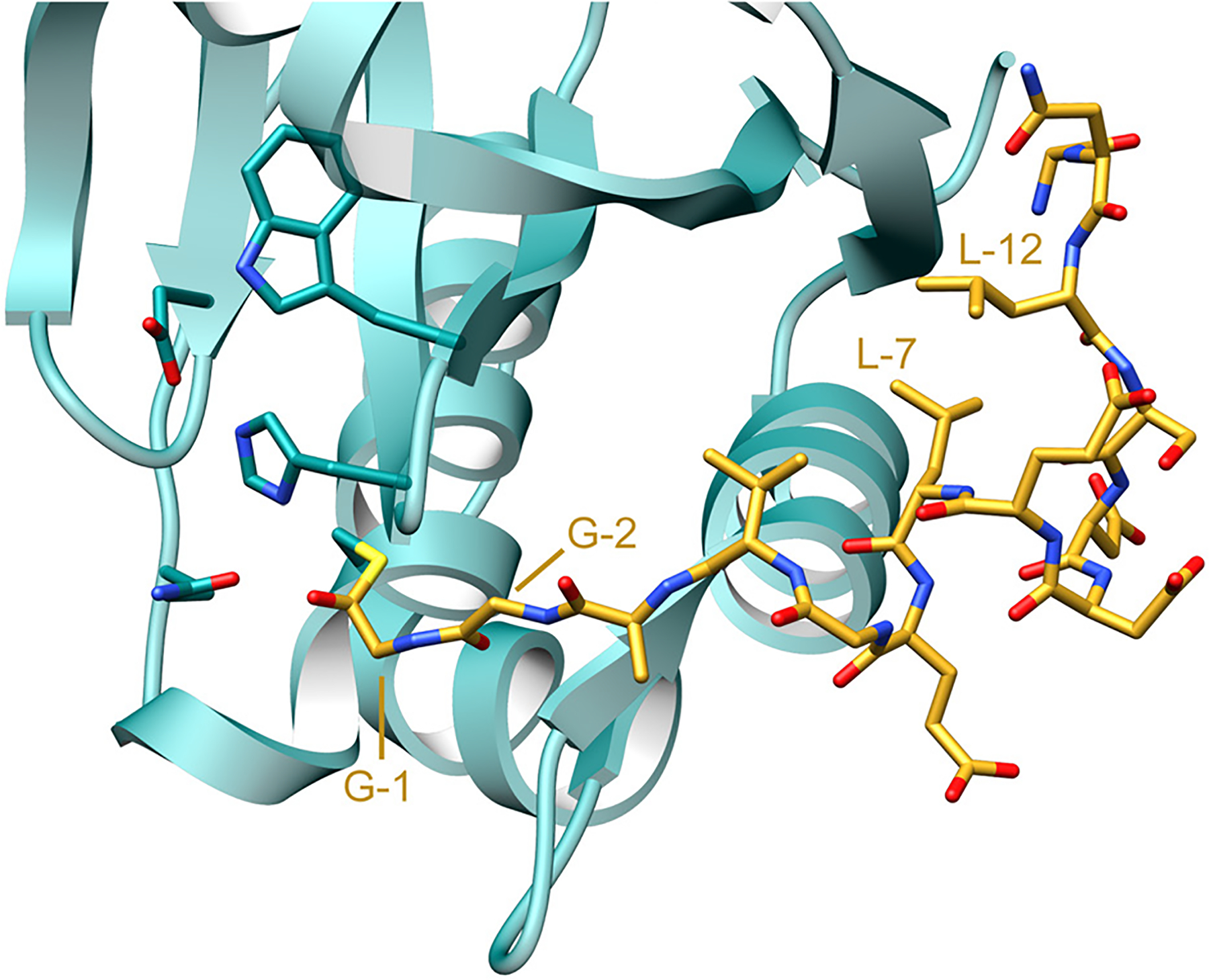
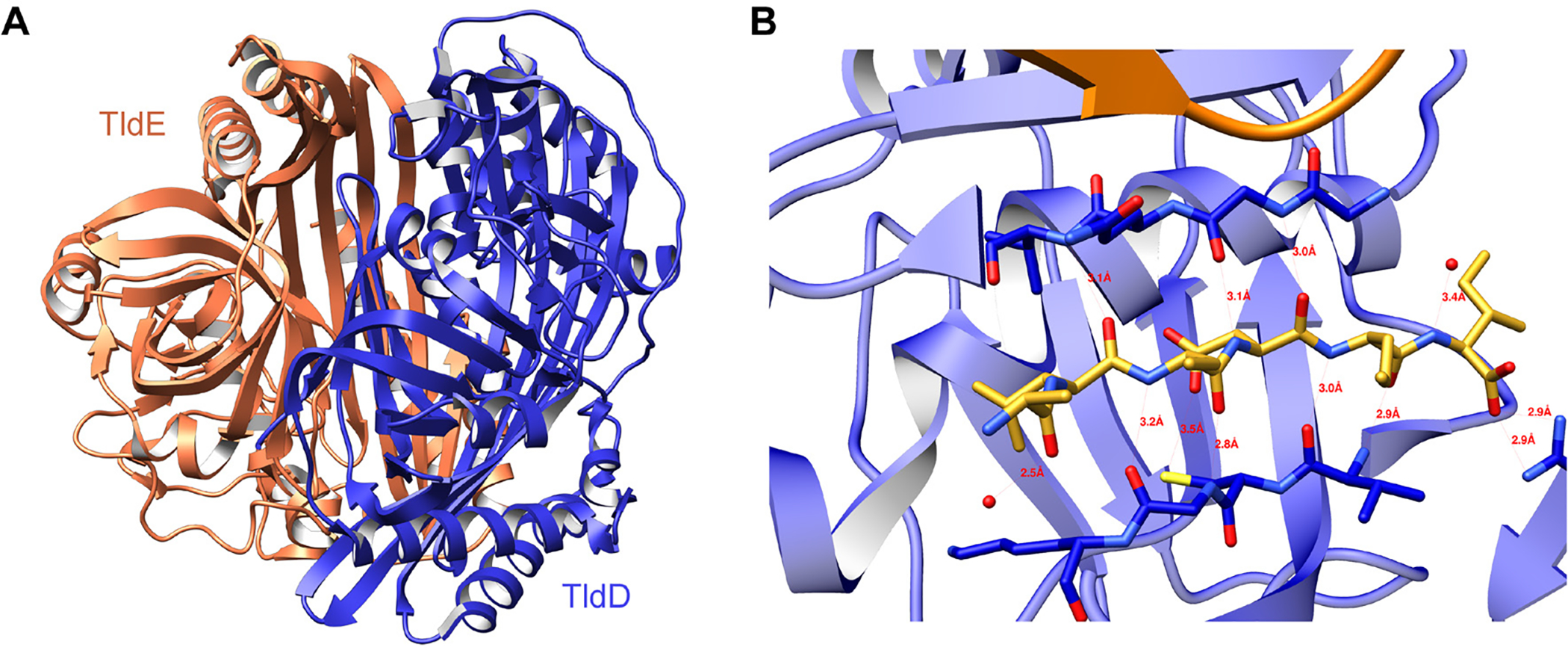
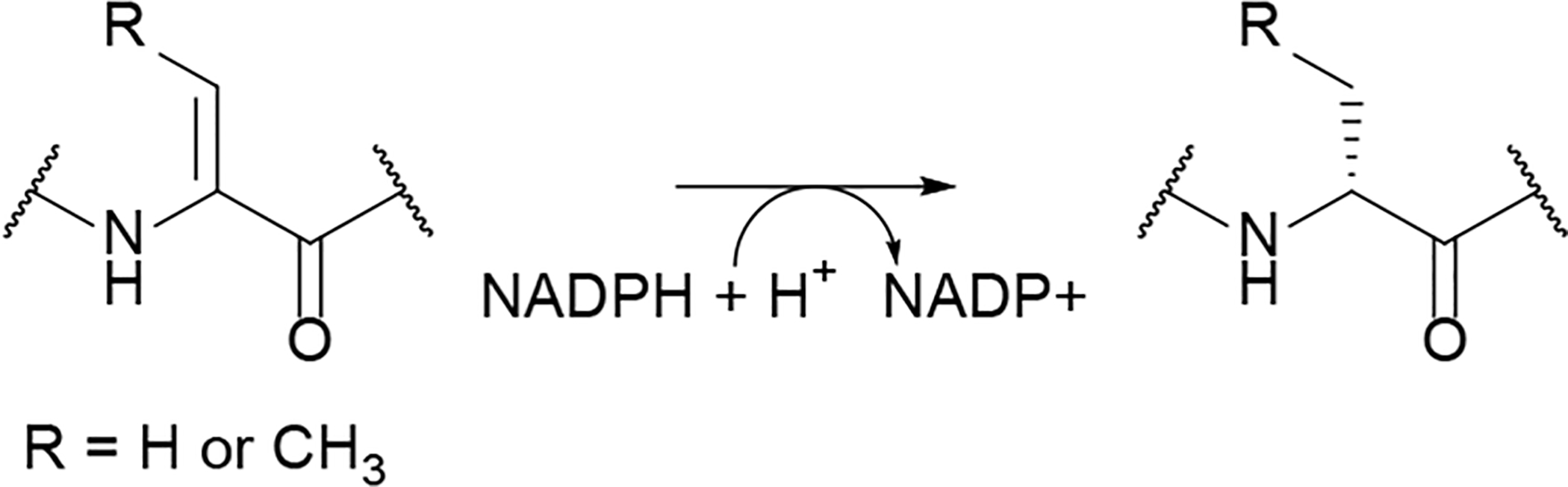
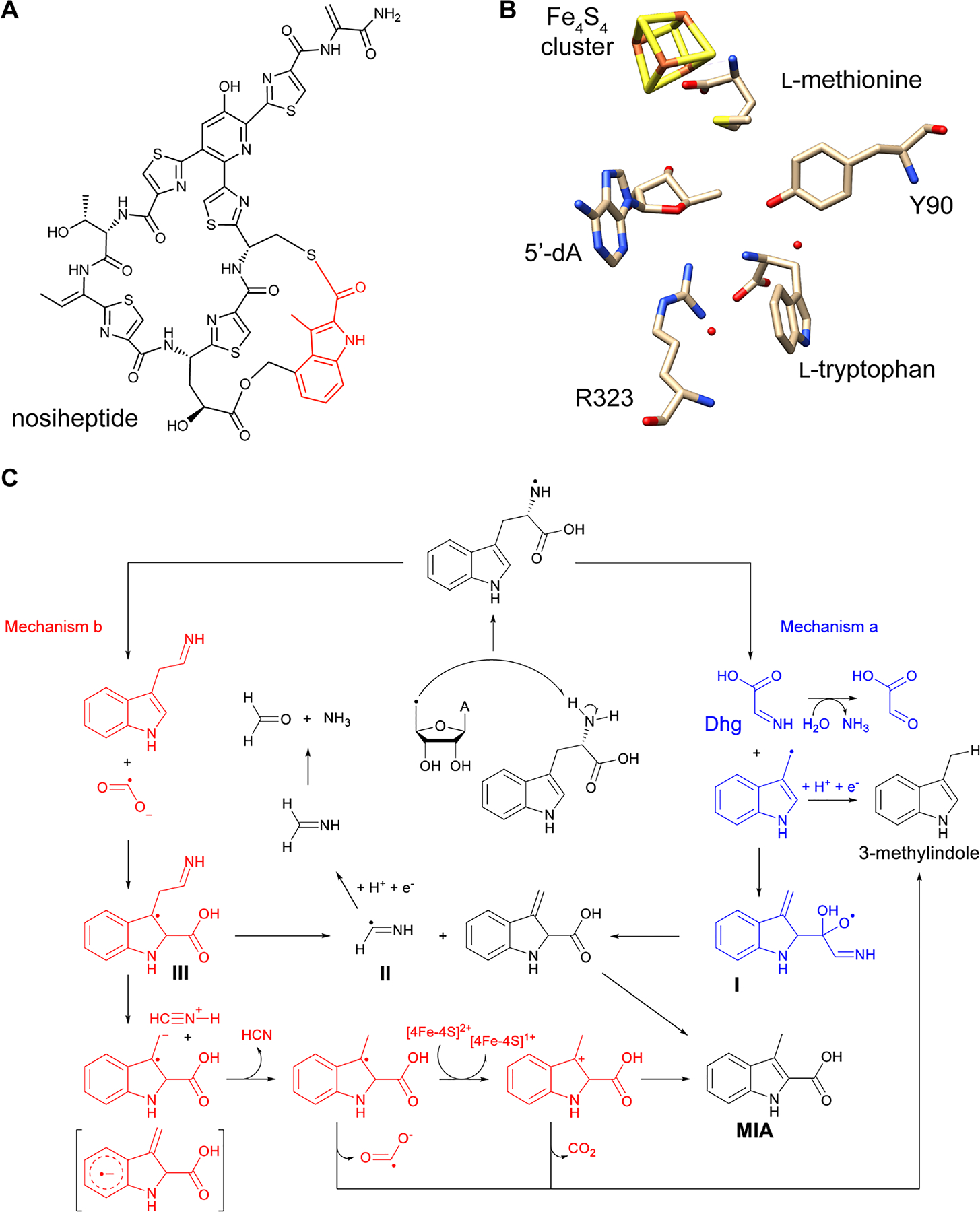
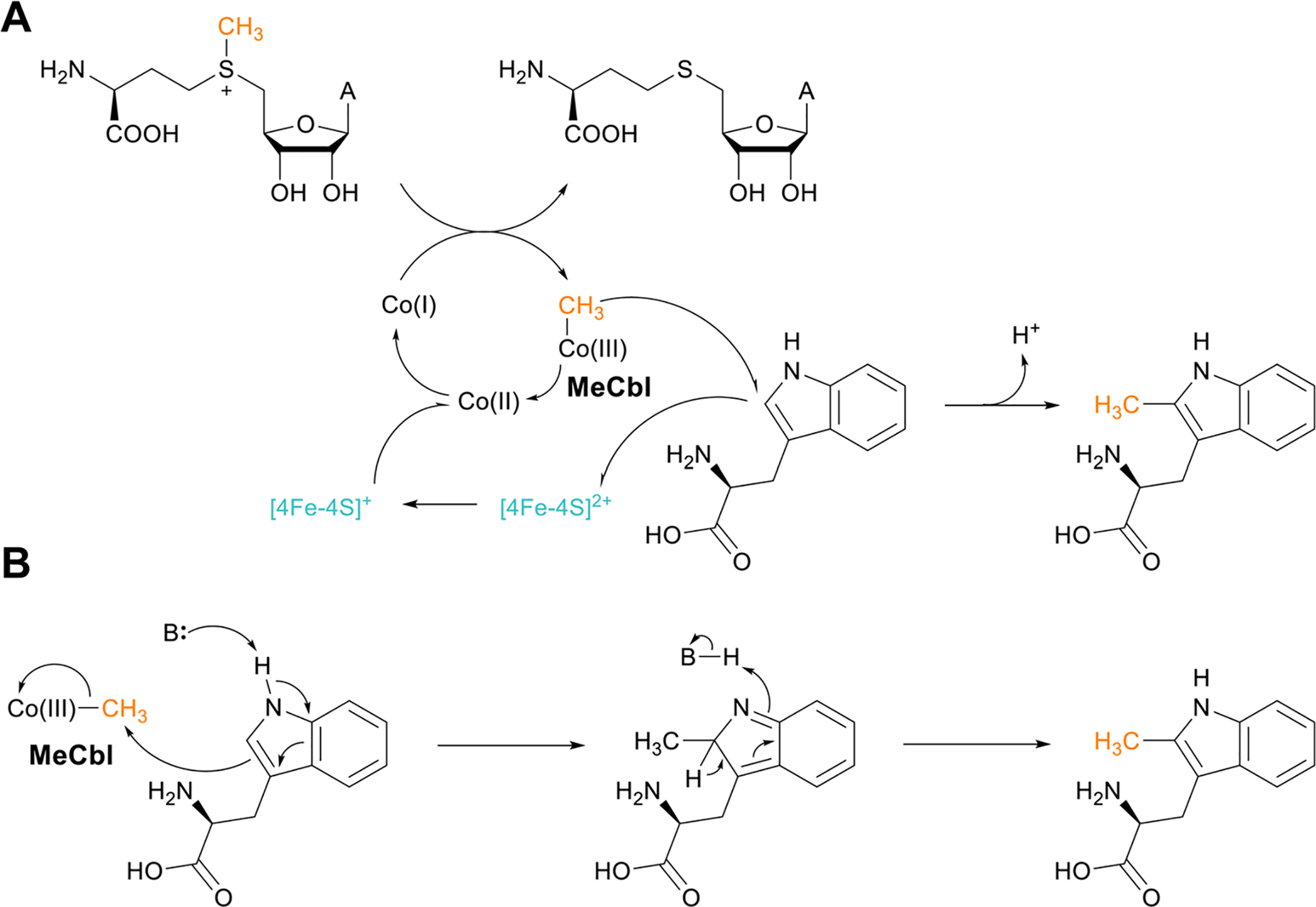
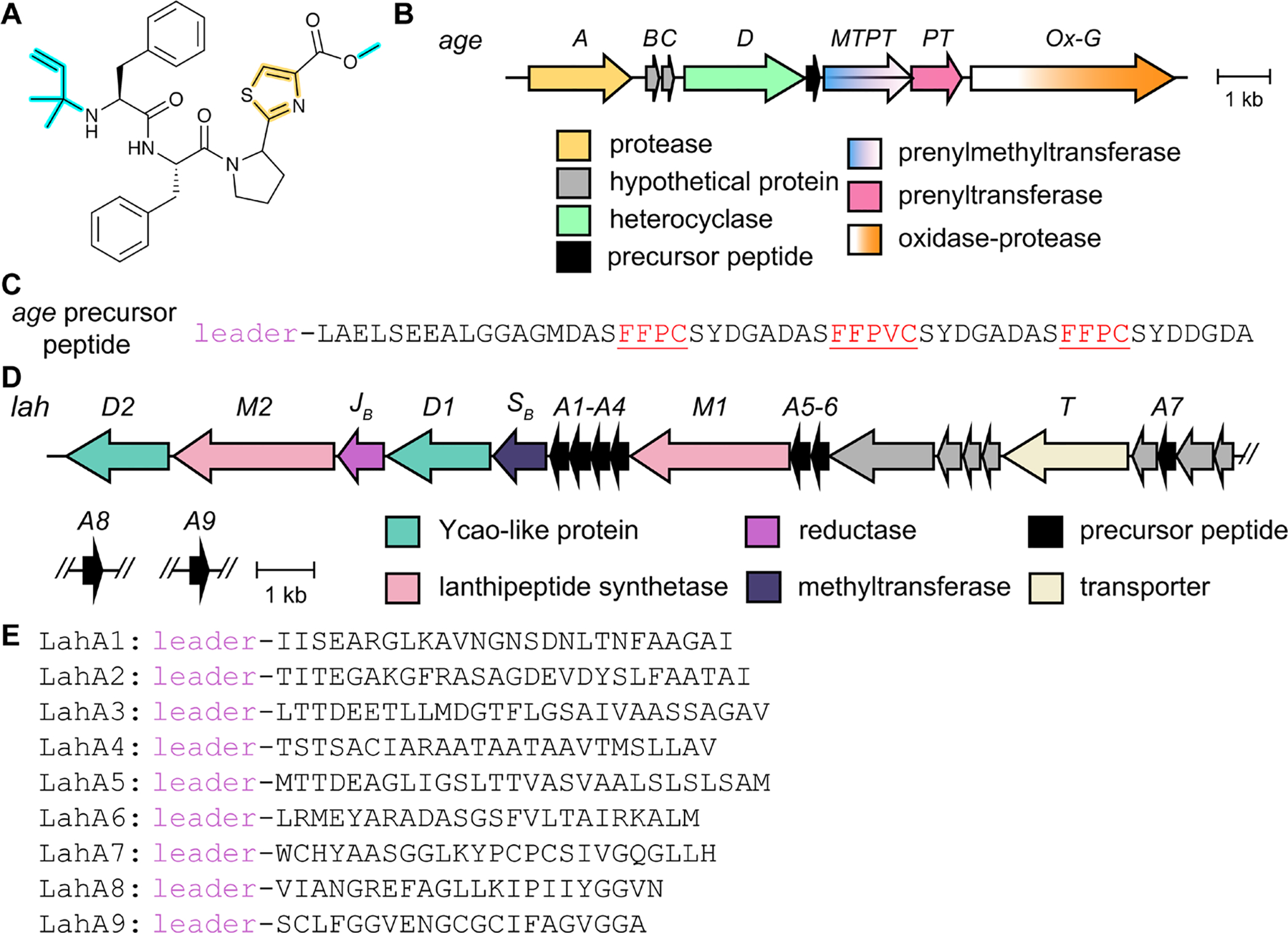
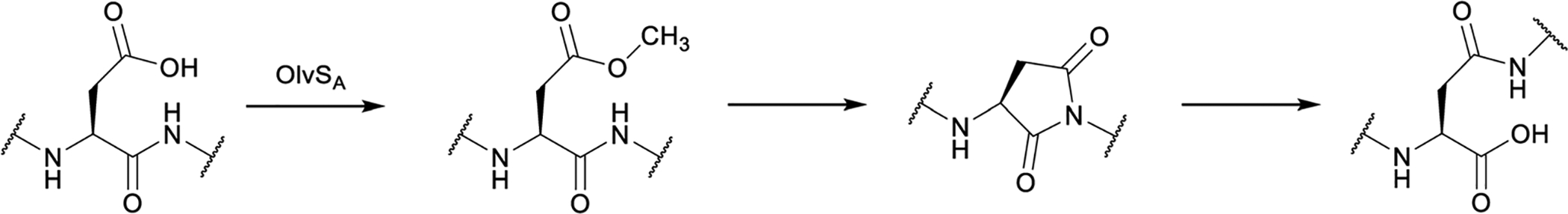
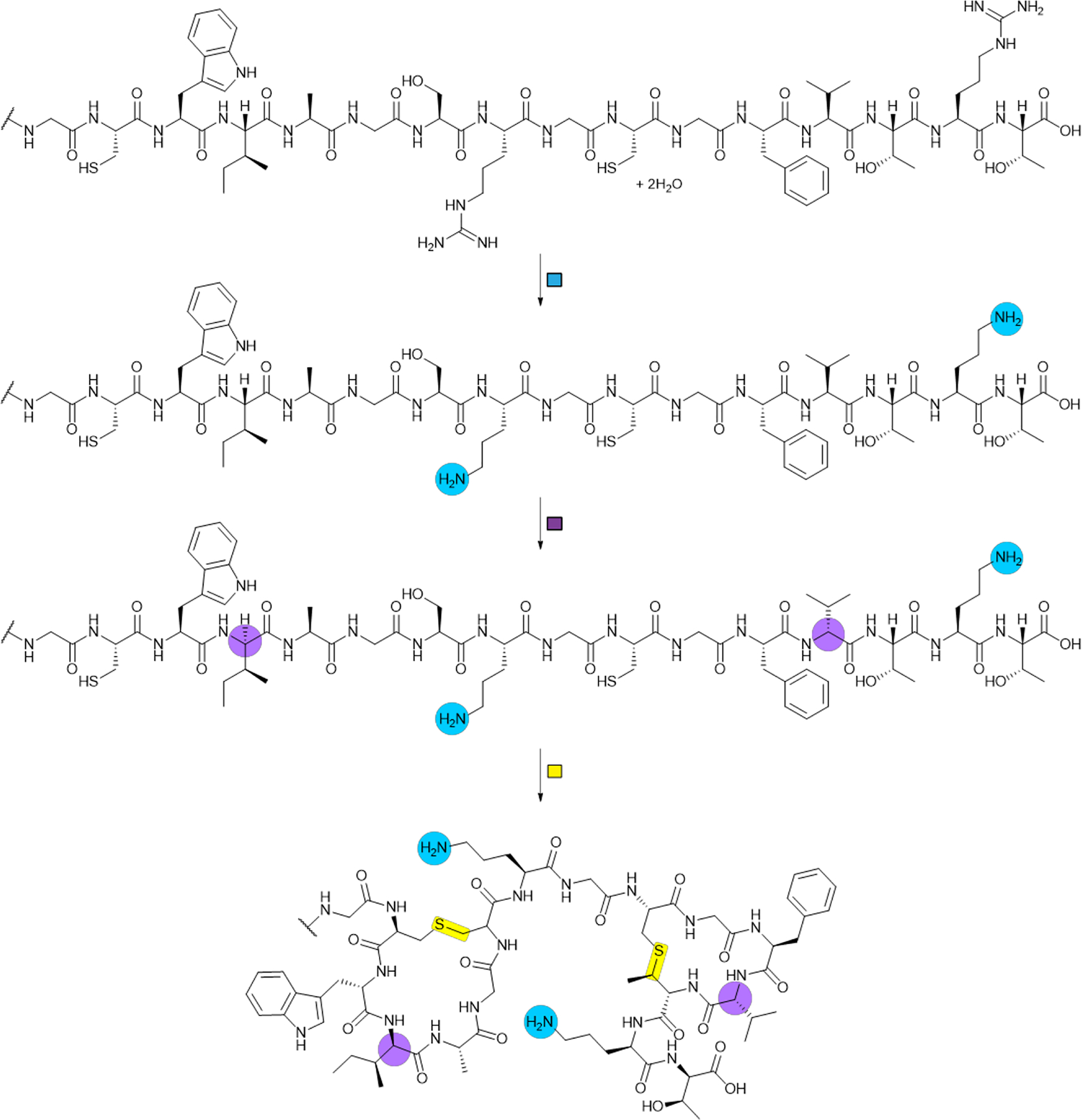
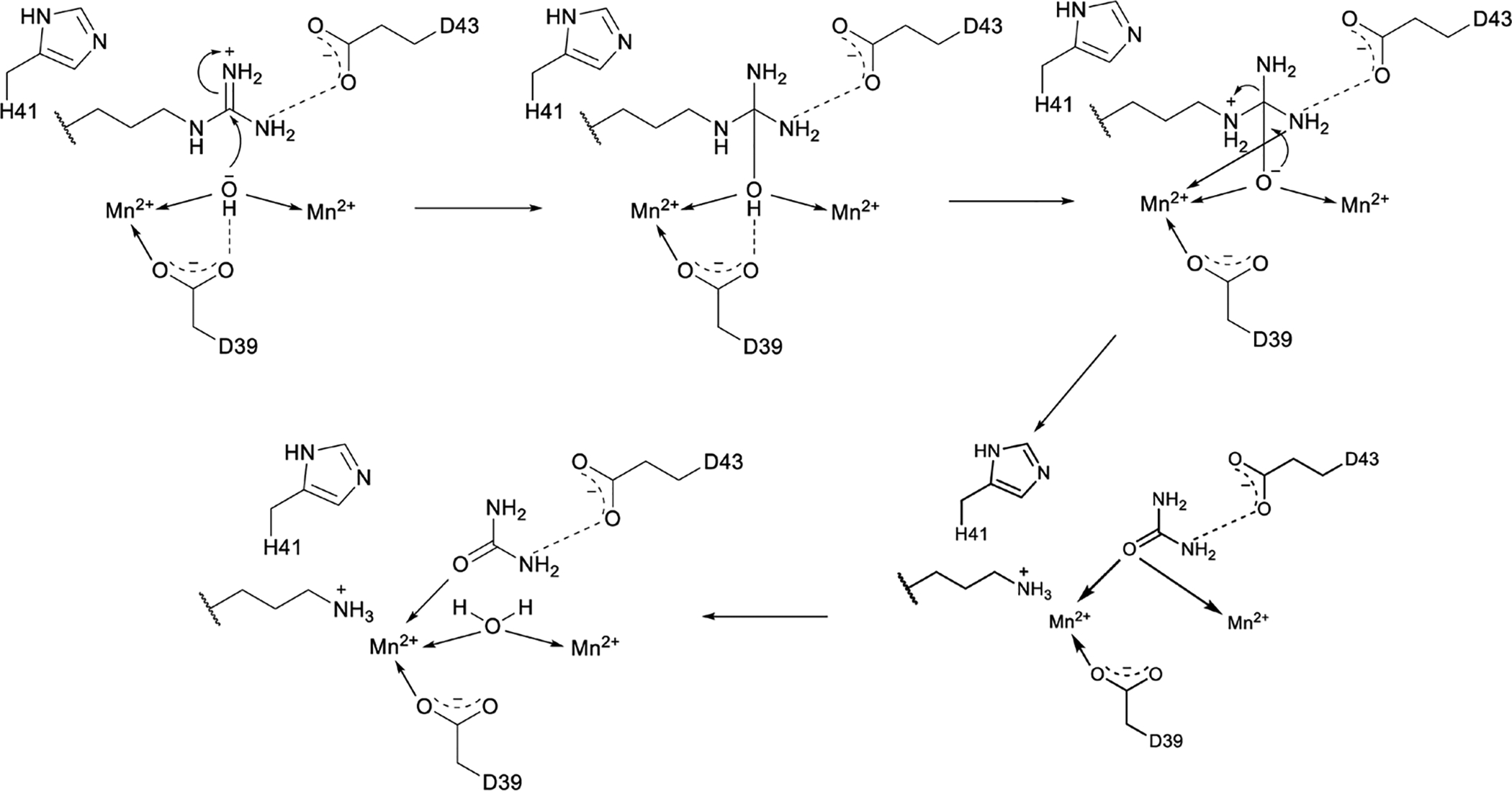
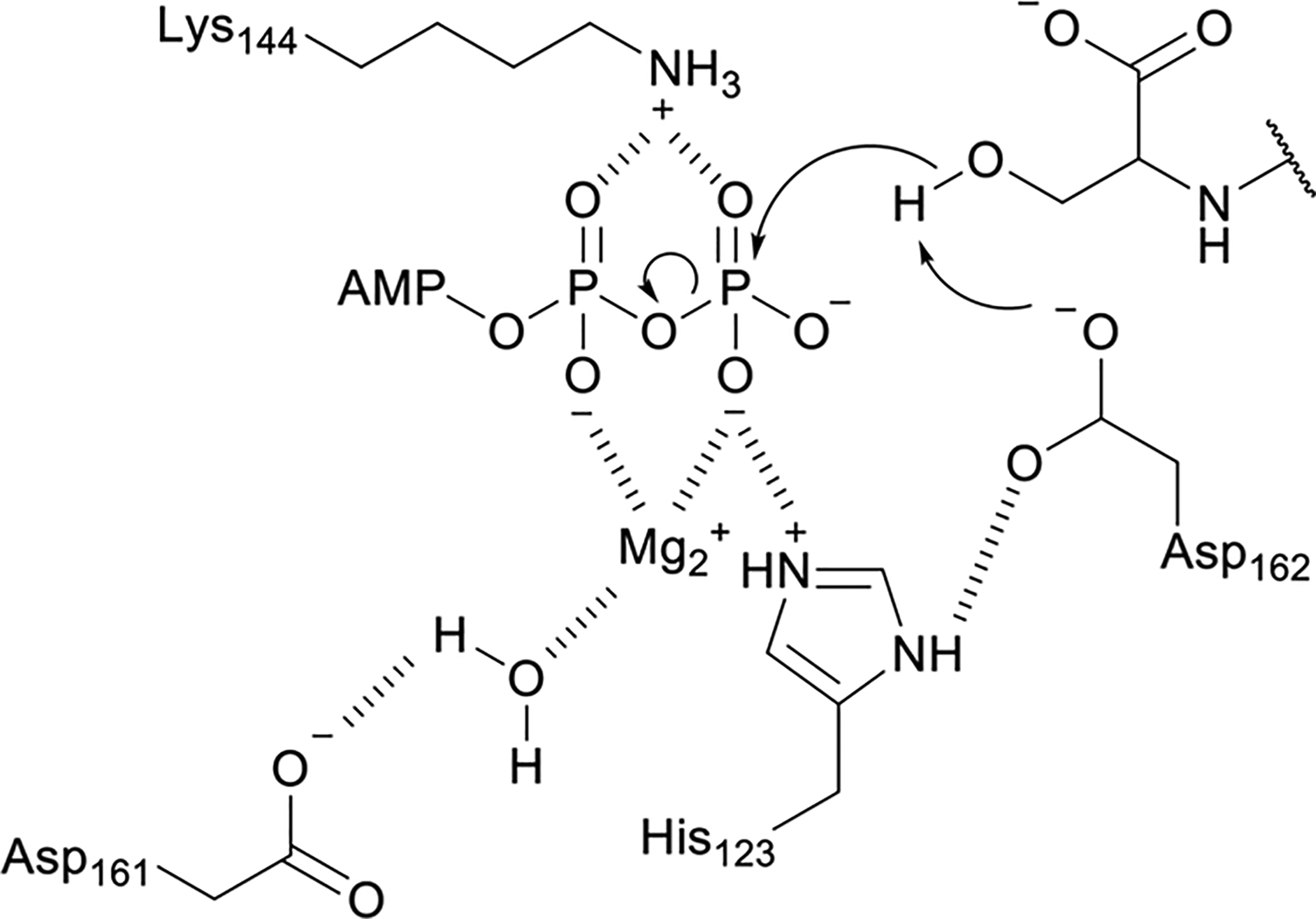
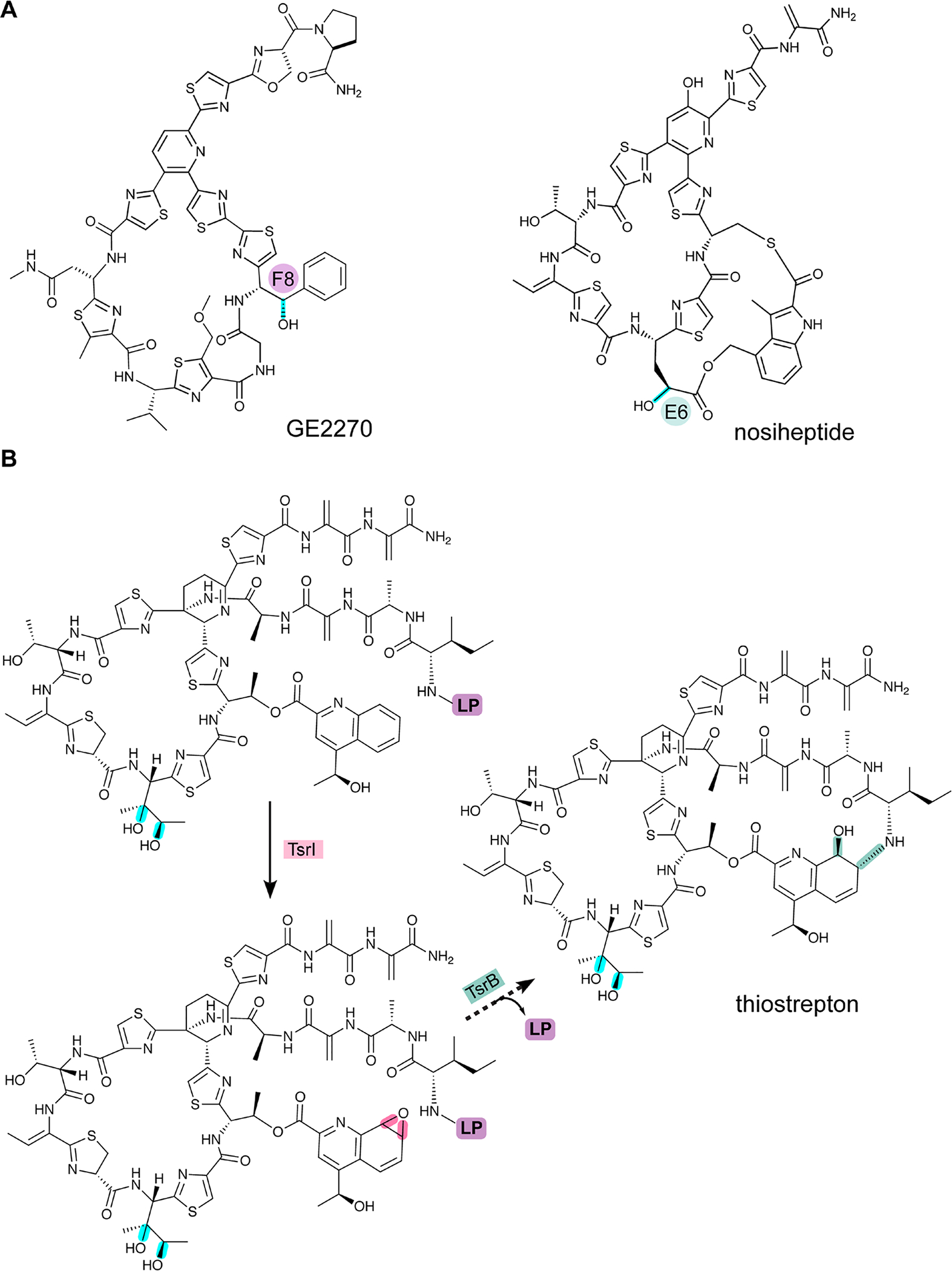
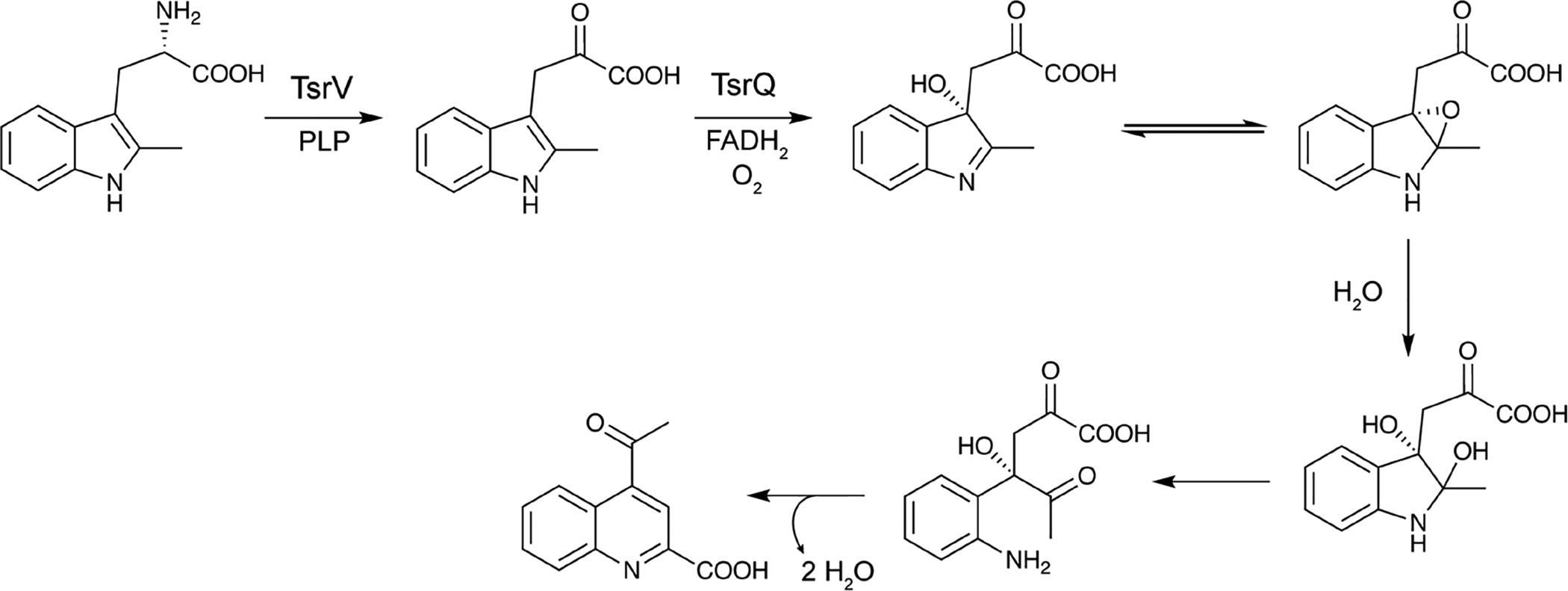
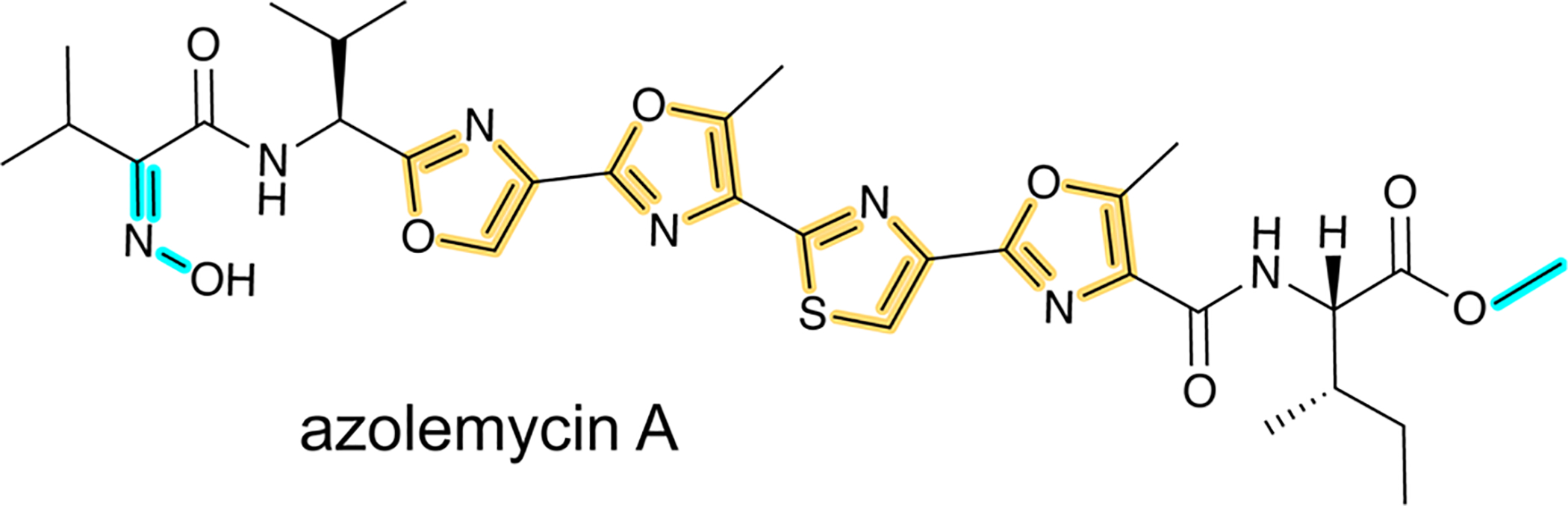
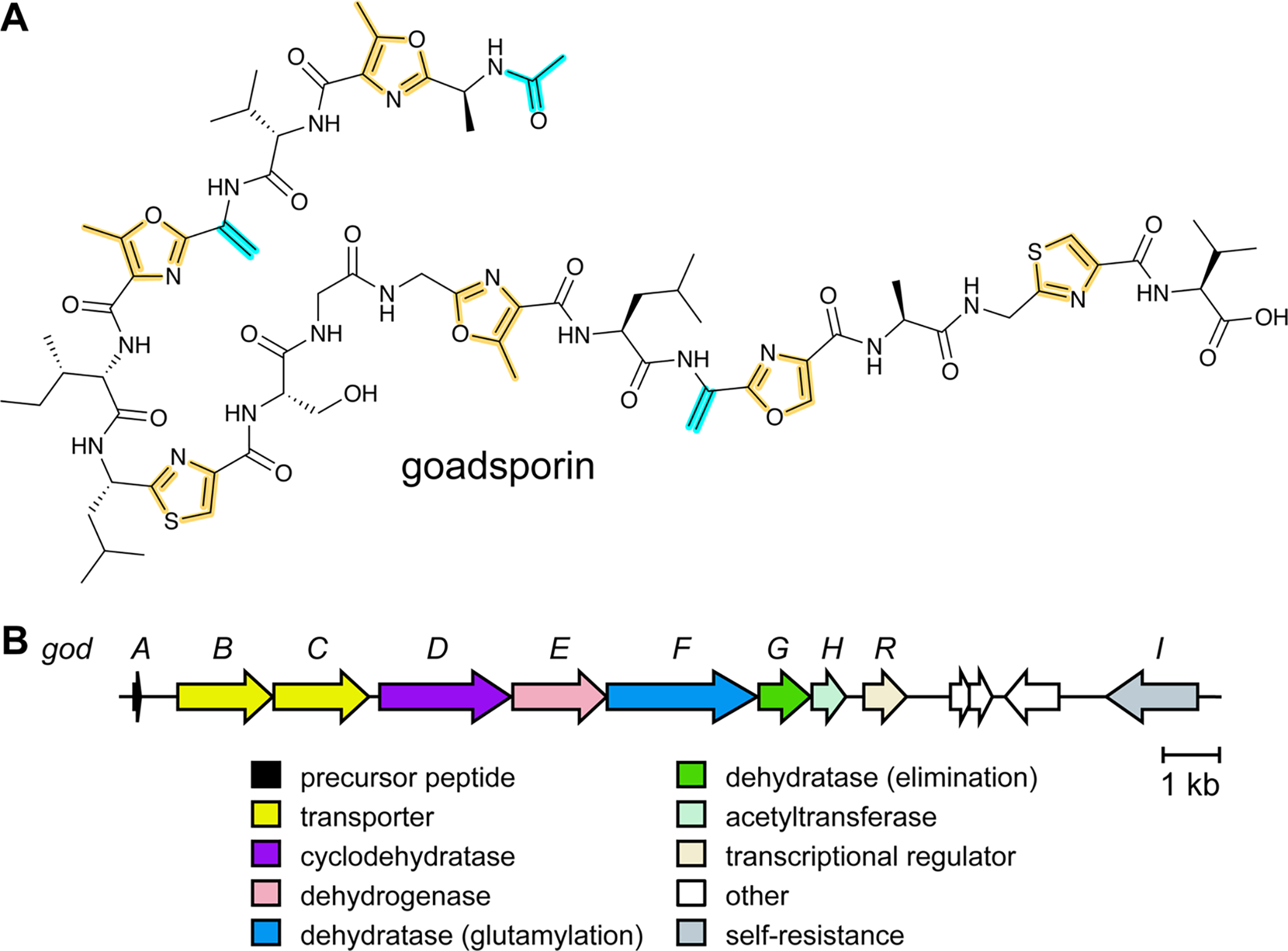
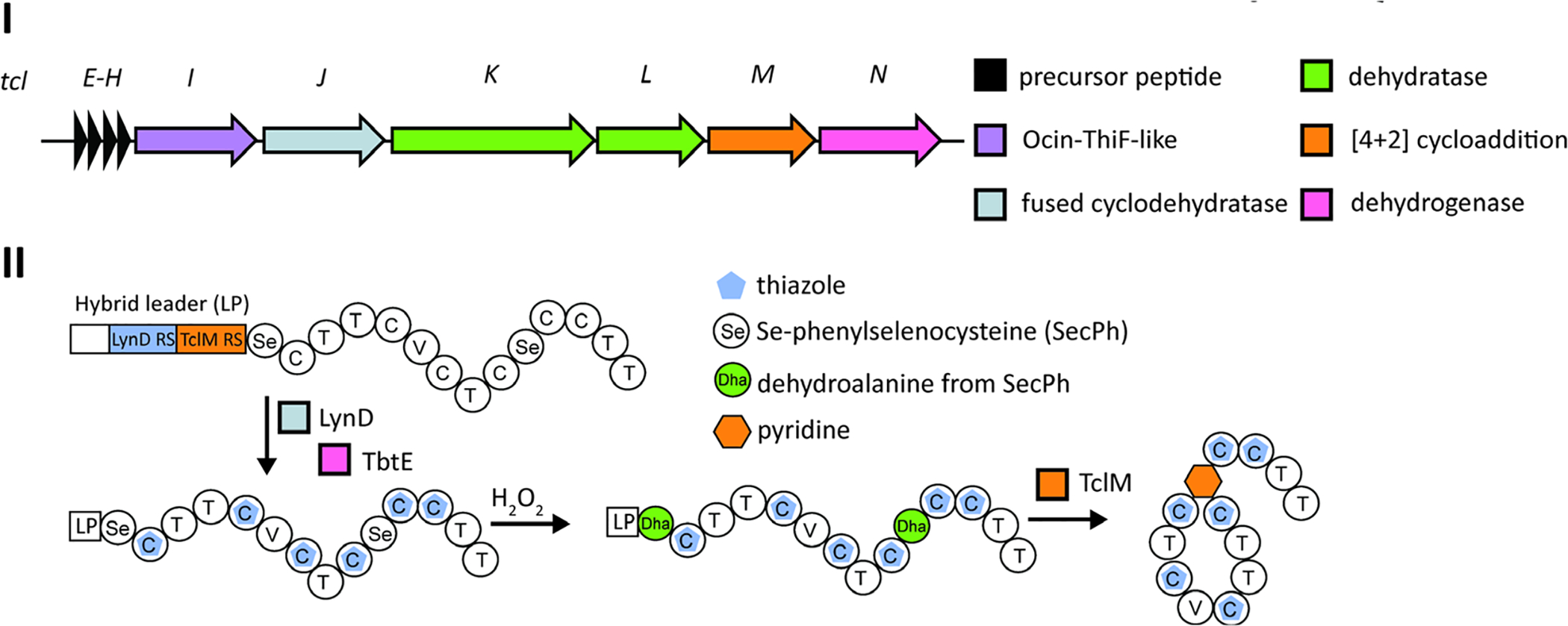
References
-
- Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian K-D, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl H-G, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang G-L, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM and van der Donk WA, Nat. Prod. Rep, 2013, 30, 108–160. - PMC - PubMed
-
- Plat A, Kuipers A, Rink R and Moll GN, Current protein & peptide science, 2013, 14, 85–96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources