

Experimental autoimmune encephalomyelitis is associated with changes of the microbiota composition in the gastrointestinal tract
- PMID: 32938979
- PMCID: PMC7494894
- DOI: 10.1038/s41598-020-72197-y
Experimental autoimmune encephalomyelitis is associated with changes of the microbiota composition in the gastrointestinal tract
Abstract
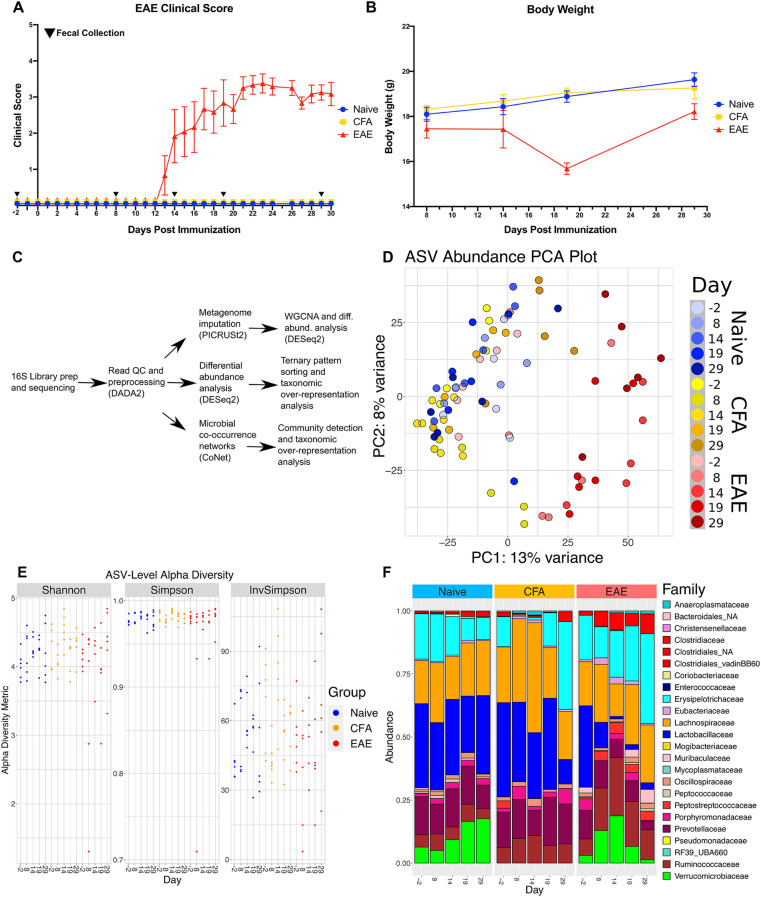
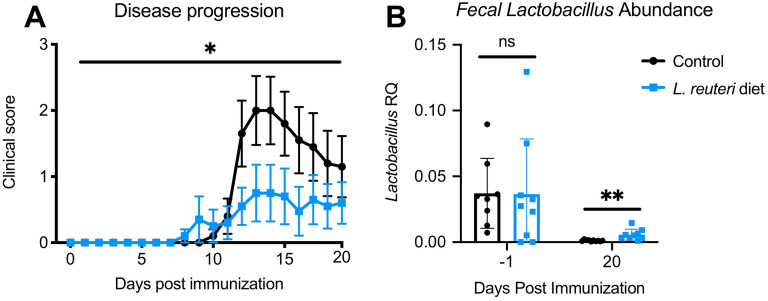
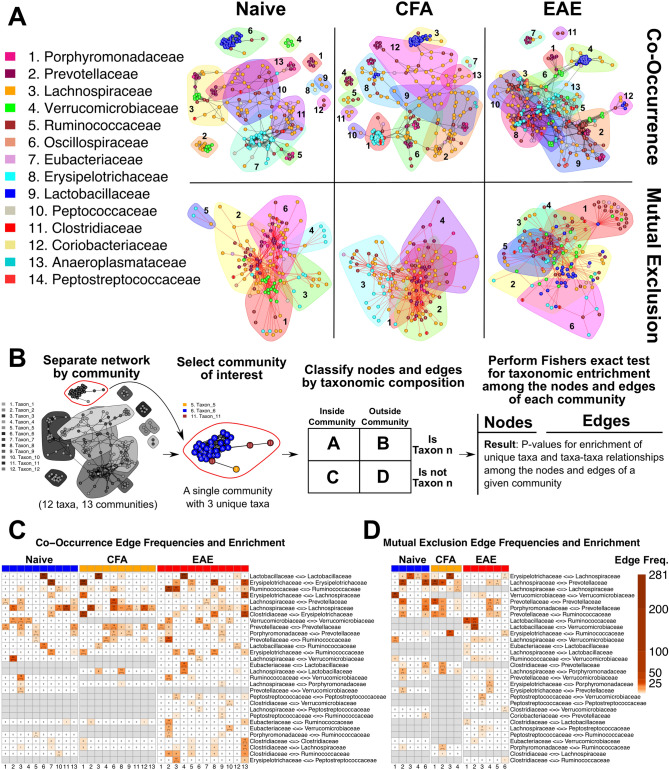
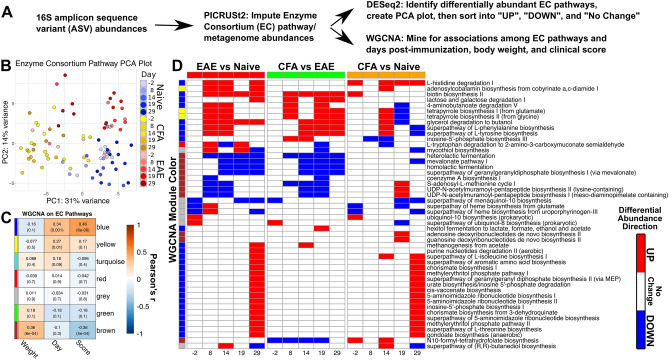
The gut microbiome is known to be sensitive to changes in the immune system, especially during autoimmune diseases such as Multiple Sclerosis (MS). Our study examines the changes to the gut microbiome that occur during experimental autoimmune encephalomyelitis (EAE), an animal model for MS. We collected fecal samples at key stages of EAE progression and quantified microbial abundances with 16S V3-V4 amplicon sequencing. Our analysis of the data suggests that the abundance of commensal Lactobacillaceae decreases during EAE while other commensal populations belonging to the Clostridiaceae, Ruminococcaceae, and Peptostreptococcaceae families expand. Community analysis with microbial co-occurrence networks points to these three expanding taxa as potential mediators of gut microbiome dysbiosis. We also employed PICRUSt2 to impute MetaCyc Enzyme Consortium (EC) pathway abundances from the original microbial abundance data. From this analysis, we found that a number of imputed EC pathways responsible for the production of immunomodulatory compounds appear to be enriched in mice undergoing EAE. Our analysis and interpretation of results provides a detailed picture of the changes to the gut microbiome that are occurring throughout the course of EAE disease progression and helps to evaluate EAE as a viable model for gut dysbiosis in MS patients.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
