

SHP2 promotes proliferation of breast cancer cells through regulating Cyclin D1 stability via the PI3K/AKT/GSK3β signaling pathway
- PMID: 32944401
- PMCID: PMC7476086
- DOI: 10.20892/j.issn.2095-3941.2020.0056
SHP2 promotes proliferation of breast cancer cells through regulating Cyclin D1 stability via the PI3K/AKT/GSK3β signaling pathway
Abstract
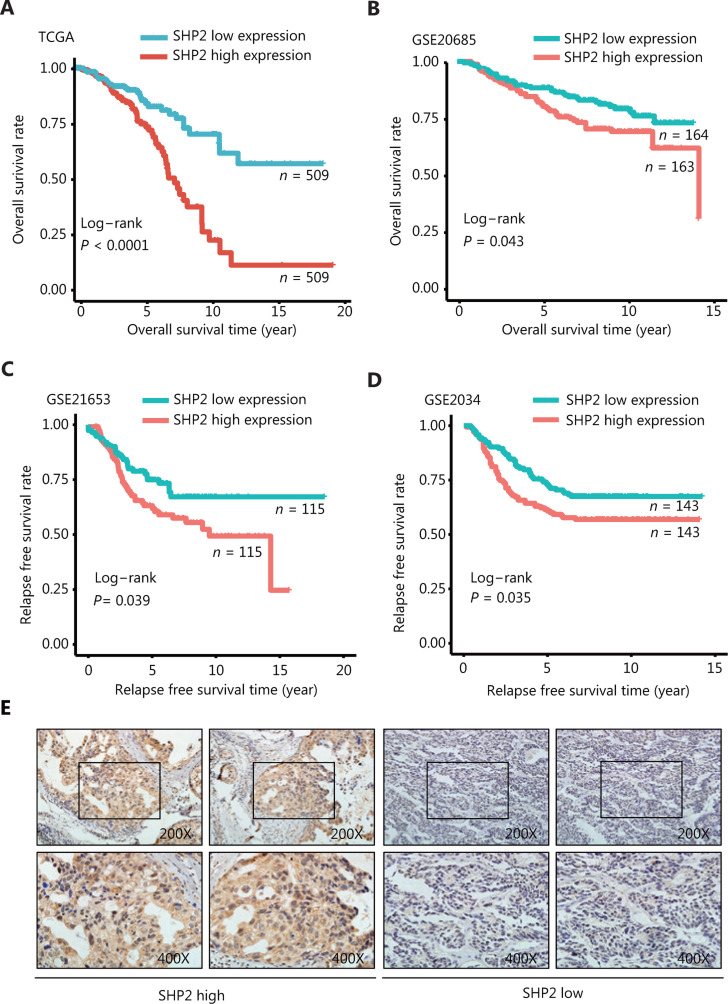
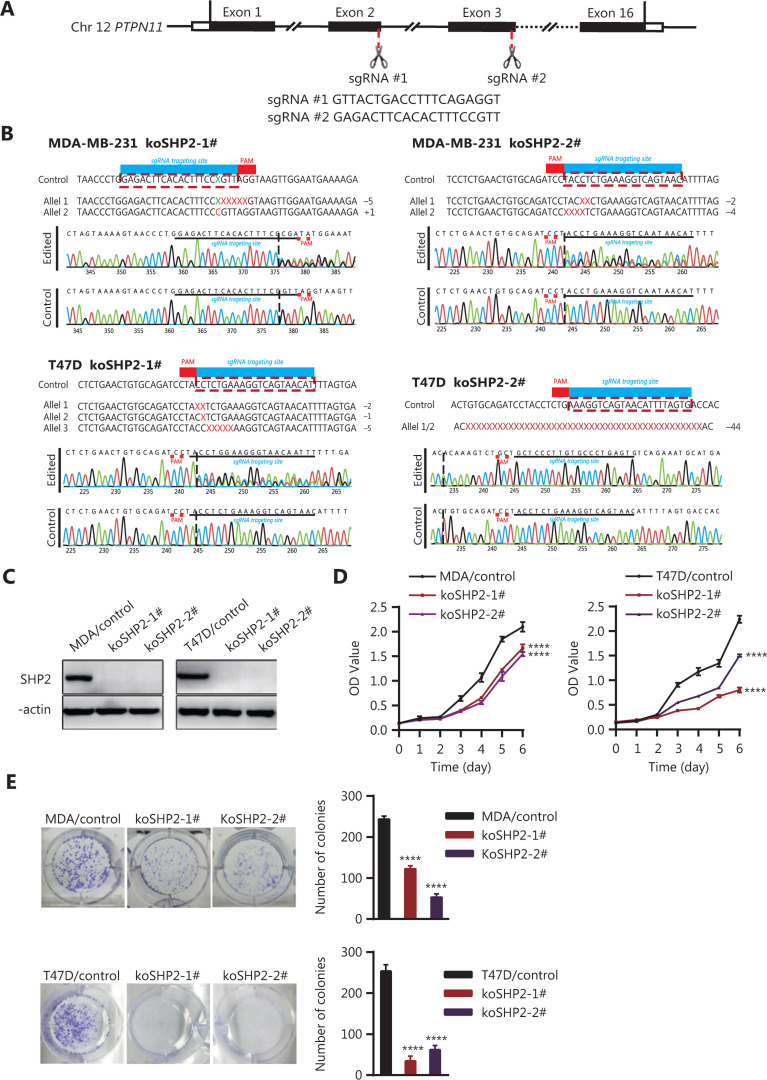
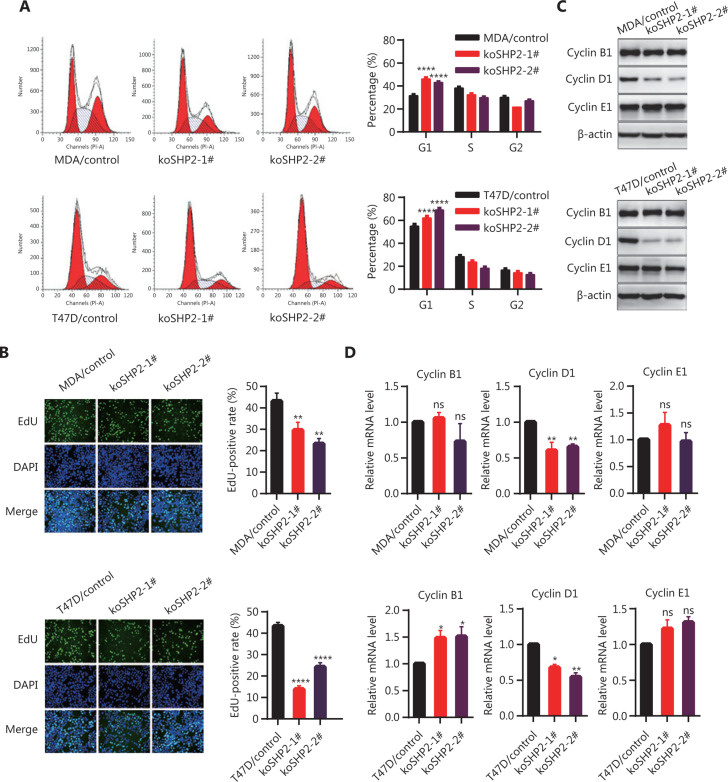
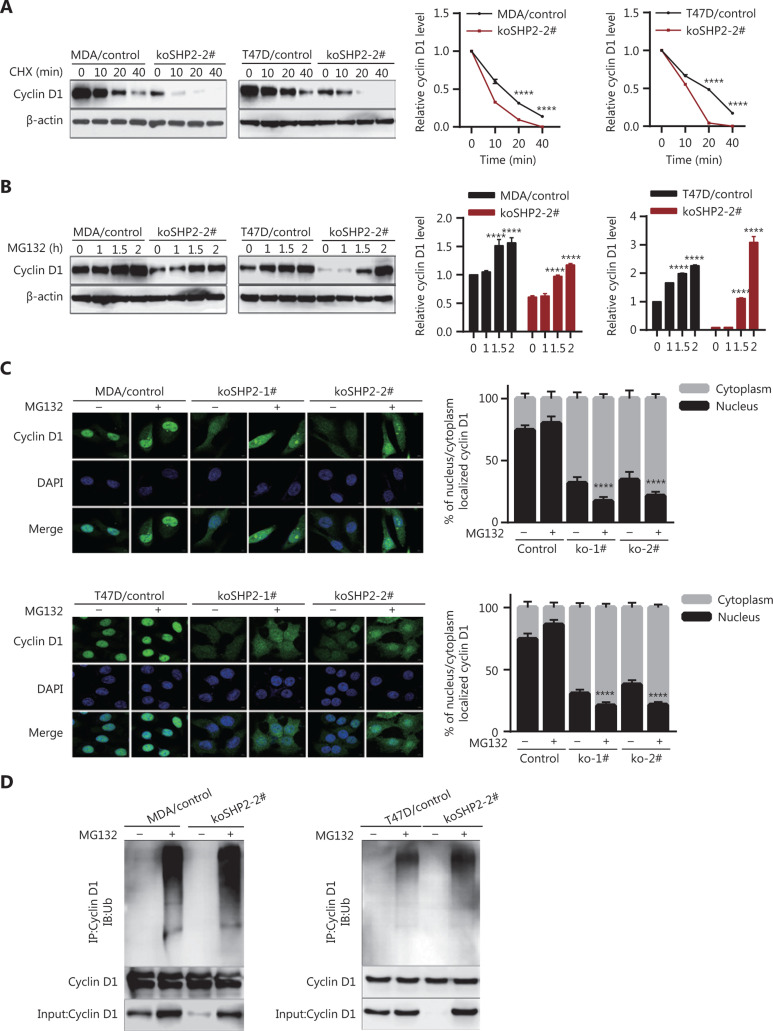
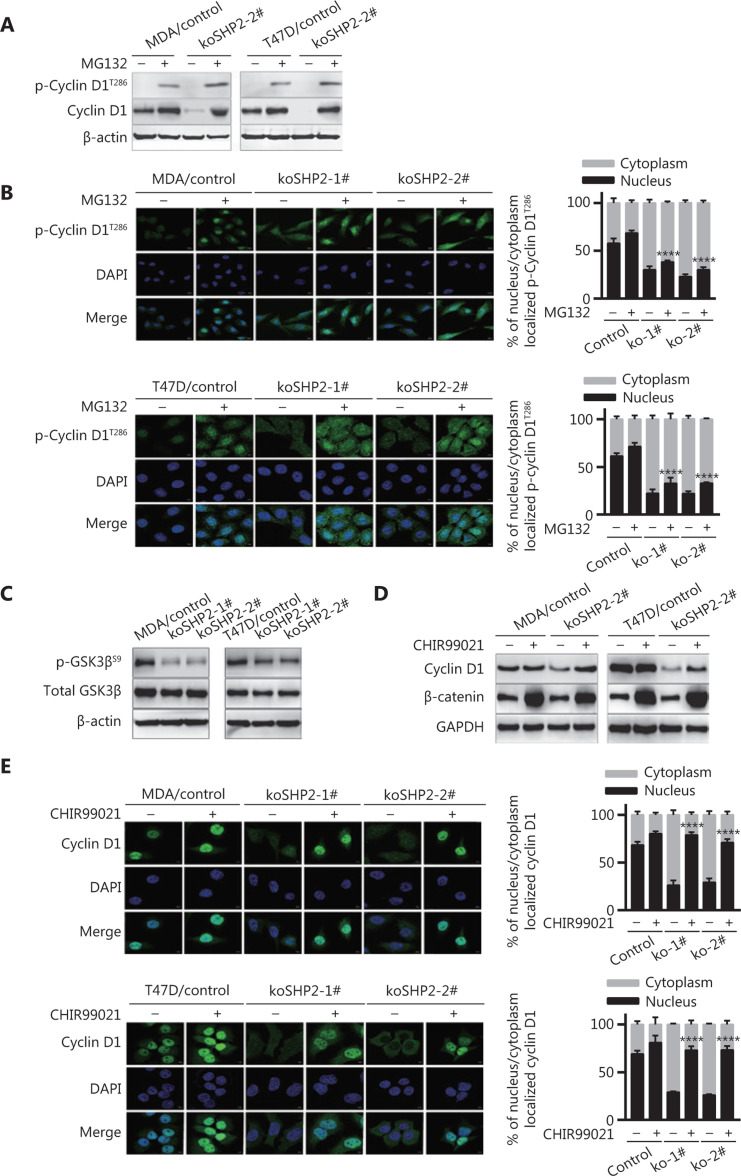
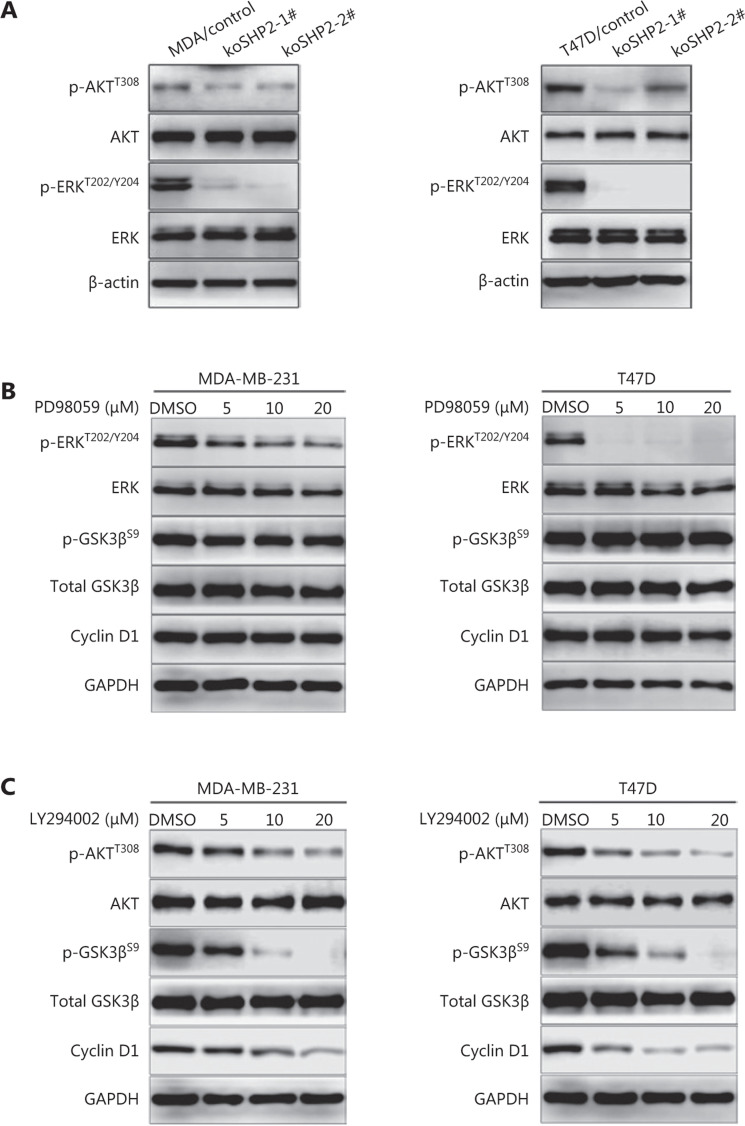
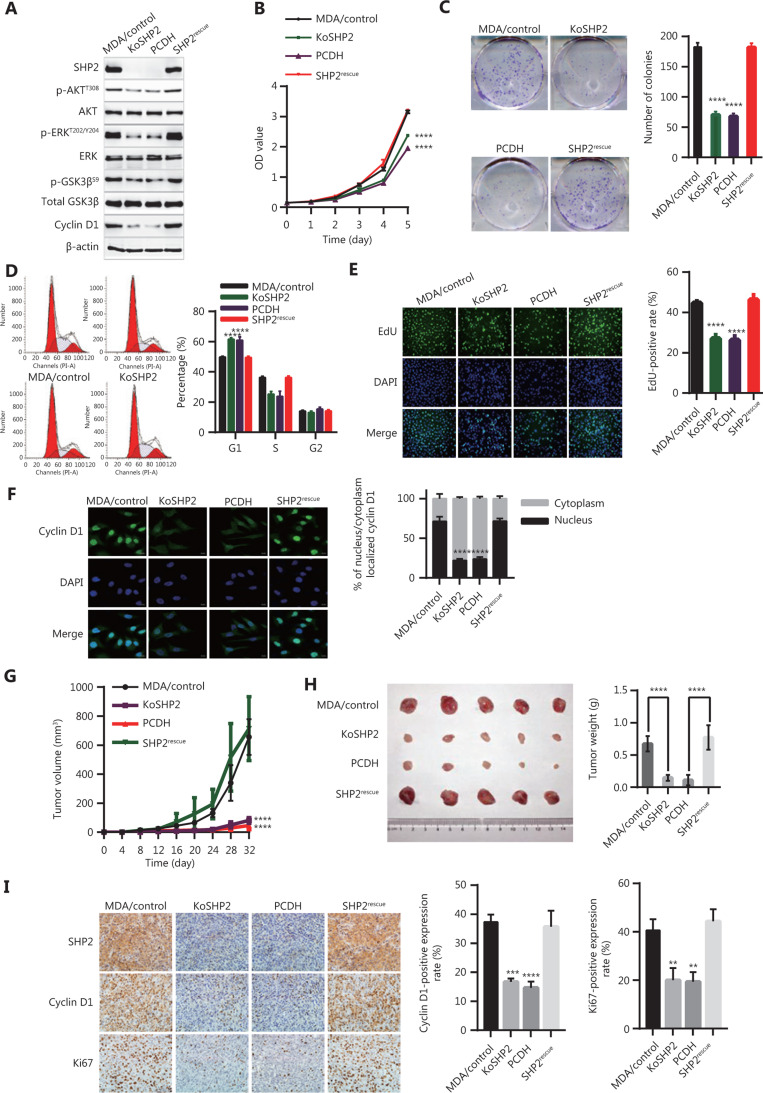
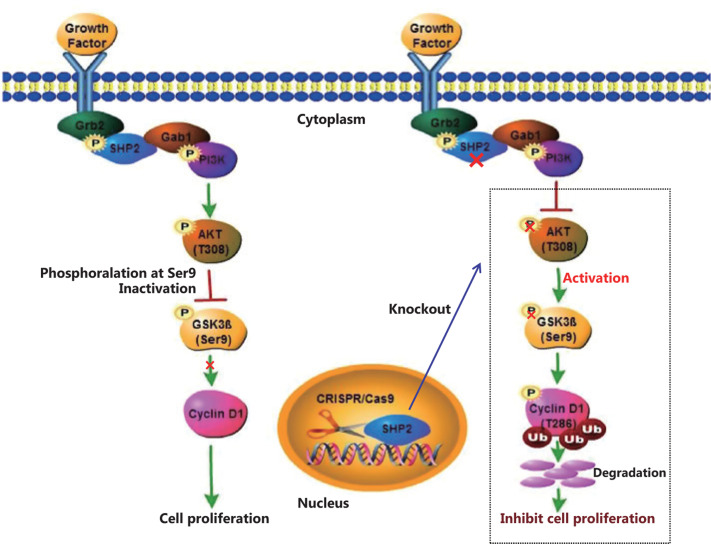
Objective: The tyrosine phosphatase SHP2 has a dual role in cancer initiation and progression in a tissue type-dependent manner. Several studies have linked SHP2 to the aggressive behavior of breast cancer cells and poorer outcomes in people with cancer. Nevertheless, the mechanistic details of how SHP2 promotes breast cancer progression remain largely undefined. Methods: The relationship between SHP2 expression and the prognosis of patients with breast cancer was investigated by using the TCGA and GEO databases. The expression of SHP2 in breast cancer tissues was analyzed by immunohistochemistry. CRISPR/Cas9 technology was used to generate SHP2-knockout breast cancer cells. Cell-counting kit-8, colony formation, cell cycle, and EdU incorporation assays, as well as a tumor xenograft model were used to examine the function of SHP2 in breast cancer proliferation. Quantitative RT-PCR, western blotting, immunofluorescence staining, and ubiquitination assays were used to explore the molecular mechanism through which SHP2 regulates breast cancer proliferation. Results: High SHP2 expression is correlated with poor prognosis in patients with breast cancer. SHP2 is required for the proliferation of breast cancer cells in vitro and tumor growth in vivo through regulation of Cyclin D1 abundance, thereby accelerating cell cycle progression. Notably, SHP2 modulates the ubiquitin-proteasome-dependent degradation of Cyclin D1 via the PI3K/AKT/GSK3β signaling pathway. SHP2 knockout attenuates the activation of PI3K/AKT signaling and causes the dephosphorylation and resultant activation of GSK3β. GSK3β then mediates phosphorylation of Cyclin D1 at threonine 286, thereby promoting the translocation of Cyclin D1 from the nucleus to the cytoplasm and facilitating Cyclin D1 degradation through the ubiquitin-proteasome system. Conclusions: Our study uncovered the mechanism through which SHP2 regulates breast cancer proliferation. SHP2 may therefore potentially serve as a therapeutic target for breast cancer.
Keywords: Cyclin D1; GSK3β; PI3K/AKT; SHP2; breast cancer; proliferation.
Copyright: © 2020, Cancer Biology & Medicine.
Conflict of interest statement
*These authors contributed equally to this work.
Figures
Similar articles
-
Tyrosine phosphatase PTPN11/SHP2 in solid tumors - bull's eye for targeted therapy?Front Immunol. 2024 Mar 5;15:1340726. doi: 10.3389/fimmu.2024.1340726. eCollection 2024. Front Immunol. 2024. PMID: 38504984 Free PMC article. Review.
-
SHP2 inhibition and adjuvant therapy synergistically target KIT-mutant GISTs via ERK1/2-regulated GSK3β/cyclin D1 pathway.Clin Transl Med. 2025 Feb;15(2):e70231. doi: 10.1002/ctm2.70231. Clin Transl Med. 2025. PMID: 39981588 Free PMC article.
-
Expression of PRIP, a phosphatidylinositol 4,5-bisphosphate binding protein, attenuates PI3K/AKT signaling and suppresses tumor growth in a xenograft mouse model.Biochem Biophys Res Commun. 2021 May 7;552:106-113. doi: 10.1016/j.bbrc.2021.03.045. Epub 2021 Mar 18. Biochem Biophys Res Commun. 2021. PMID: 33743346
-
Overexpression of IC53d promotes the proliferation of gastric cancer cells by activating the AKT/GSK3β/cyclin D1 signaling pathway.Oncol Rep. 2019 May;41(5):2739-2752. doi: 10.3892/or.2019.7042. Epub 2019 Mar 5. Oncol Rep. 2019. PMID: 30864700 Free PMC article.
-
A comprehensive review of SHP2 and its role in cancer.Cell Oncol (Dordr). 2022 Oct;45(5):729-753. doi: 10.1007/s13402-022-00698-1. Epub 2022 Sep 6. Cell Oncol (Dordr). 2022. PMID: 36066752 Review.
Cited by
-
PTPN11 is a potential biomarker for type 2 diabetes mellitus complicated with colorectal cancer.Sci Rep. 2024 Oct 24;14(1):25155. doi: 10.1038/s41598-024-75889-x. Sci Rep. 2024. PMID: 39448762 Free PMC article.
-
The Toxoplasma secreted effector TgWIP modulates dendritic cell motility by activating host tyrosine phosphatases Shp1 and Shp2.Res Sq [Preprint]. 2024 Jun 26:rs.3.rs-4539584. doi: 10.21203/rs.3.rs-4539584/v1. Res Sq. 2024. Update in: Cell Mol Life Sci. 2024 Jul 9;81(1):294. doi: 10.1007/s00018-024-05283-3. PMID: 38978596 Free PMC article. Updated. Preprint.
-
Mitochondrial Breast Cancer Resistant Protein Sustains the Proliferation and Survival of Drug-Resistant Breast Cancer Cells by Regulating Intracellular Reactive Oxygen Species.Front Cell Dev Biol. 2021 Sep 28;9:719209. doi: 10.3389/fcell.2021.719209. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34650973 Free PMC article.
-
Tyrosine phosphatase PTPN11/SHP2 in solid tumors - bull's eye for targeted therapy?Front Immunol. 2024 Mar 5;15:1340726. doi: 10.3389/fimmu.2024.1340726. eCollection 2024. Front Immunol. 2024. PMID: 38504984 Free PMC article. Review.
-
Tumor-associated macrophages derived exosomal circPLK1 promotes resistance to EGFR inhibitor osimertinib in non-small cell lung cancer.Discov Oncol. 2025 Jul 1;16(1):1196. doi: 10.1007/s12672-025-03025-w. Discov Oncol. 2025. PMID: 40591172 Free PMC article.
References
-
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. - PubMed
-
- Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32. - PubMed
-
- Rapino F, Delaunay S, Rambow F, Zhou Z, Tharun L, De Tullio P, et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature. 2018;558:605–9. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous