

Revisiting the PD-1 pathway
- PMID: 32948597
- PMCID: PMC7500922
- DOI: 10.1126/sciadv.abd2712
Revisiting the PD-1 pathway
Abstract
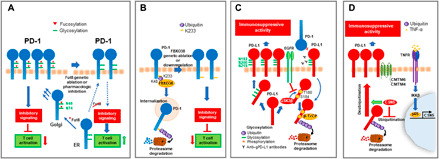
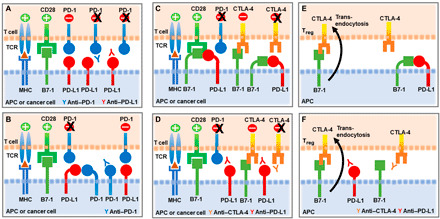
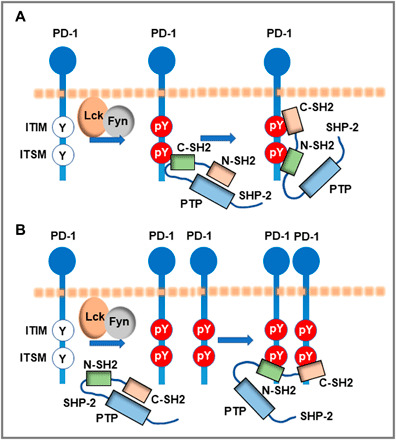
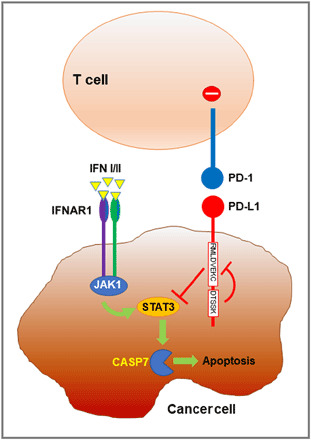
Programmed Death-1 (PD-1; CD279) is an inhibitory receptor induced in activated T cells. PD-1 engagement by its ligands, PD-L1 and PD-L2, maintains peripheral tolerance but also compromises anti-tumor immunity. Blocking antibodies against PD-1 or its ligands have revolutionized cancer immunotherapy. However, only a fraction of patients develop durable antitumor responses. Clinical outcomes have reached a plateau without substantial advances by combinatorial approaches. Thus, great interest has recently emerged to investigate, in depth, the mechanisms by which the PD-1 pathway transmits inhibitory signals with the goal to identify molecular targets for improvement of the therapeutic success. These efforts have revealed unpredictable dimensions of the pathway and uncovered novel mechanisms involved in PD-1 and PD-L1 regulation and function. Here, we provide an overview of the recent advances on the mechanistic aspects of the PD-1 pathway and discuss the implications of these new discoveries and the gaps that remain to be filled.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
Similar articles
-
The PD-1 Interactome.Adv Biol (Weinh). 2021 Sep;5(9):e2100758. doi: 10.1002/adbi.202100758. Epub 2021 Jun 25. Adv Biol (Weinh). 2021. PMID: 34170628 Free PMC article. Review.
-
Effects of PD-1 Signaling on Immunometabolic Reprogramming.Immunometabolism. 2022;4(2):e220007. doi: 10.20900/immunometab20220007. Epub 2022 Mar 10. Immunometabolism. 2022. PMID: 35371563 Free PMC article.
-
The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation.Front Immunol. 2016 Dec 12;7:550. doi: 10.3389/fimmu.2016.00550. eCollection 2016. Front Immunol. 2016. PMID: 28018338 Free PMC article. Review.
-
Cancer Cell-Intrinsic PD-1 and Implications in Combinatorial Immunotherapy.Front Immunol. 2018 Jul 30;9:1774. doi: 10.3389/fimmu.2018.01774. eCollection 2018. Front Immunol. 2018. PMID: 30105035 Free PMC article. Review.
-
An optimized protocol for the in vitro generation and functional analysis of human PD1/PD-L1 signal.J Recept Signal Transduct Res. 2018 Feb;38(1):31-36. doi: 10.1080/10799893.2017.1414843. Epub 2017 Dec 18. J Recept Signal Transduct Res. 2018. PMID: 29252078
Cited by
-
Multiplexed imaging mass cytometry reveals distinct tumor-immune microenvironments linked to immunotherapy responses in melanoma.Commun Med (Lond). 2022 Oct 21;2:131. doi: 10.1038/s43856-022-00197-2. eCollection 2022. Commun Med (Lond). 2022. PMID: 36281356 Free PMC article.
-
High-potency PD-1/PD-L1 degradation induced by Peptide-PROTAC in human cancer cells.Cell Death Dis. 2022 Nov 4;13(11):924. doi: 10.1038/s41419-022-05375-7. Cell Death Dis. 2022. PMID: 36333311 Free PMC article. No abstract available.
-
Attenuated Salmonella potentiate PD-L1 blockade immunotherapy in a preclinical model of colorectal cancer.Front Immunol. 2022 Dec 20;13:1017780. doi: 10.3389/fimmu.2022.1017780. eCollection 2022. Front Immunol. 2022. PMID: 36605208 Free PMC article.
-
The Emerging Interplay Between Recirculating and Tissue-Resident Memory T Cells in Cancer Immunity: Lessons Learned From PD-1/PD-L1 Blockade Therapy and Remaining Gaps.Front Immunol. 2021 Nov 16;12:755304. doi: 10.3389/fimmu.2021.755304. eCollection 2021. Front Immunol. 2021. PMID: 34867987 Free PMC article. Review.
-
The foundations of immune checkpoint blockade and the ipilimumab approval decennial.Nat Rev Drug Discov. 2022 Jul;21(7):509-528. doi: 10.1038/s41573-021-00345-8. Epub 2021 Dec 22. Nat Rev Drug Discov. 2022. PMID: 34937915 Review.
References
-
- Agata Y., Kawasaki A., Nishimura H., Ishida Y., Tsubata T., Yagita H., Honjo T., Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8, 765–772 (1996). - PubMed
-
- Nishimura H., Nose M., Hiai H., Minato N., Honjo T., Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151 (1999). - PubMed
-
- Nishimura H., Okazaki T., Tanaka Y., Nakatani K., Hara M., Matsumori A., Sasayama S., Mizoguchi A., Hiai H., Minato N., Honjo T., Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291, 319–322 (2001). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
