

NLRP3 inflammasome in endothelial dysfunction
- PMID: 32948742
- PMCID: PMC7501262
- DOI: 10.1038/s41419-020-02985-x
NLRP3 inflammasome in endothelial dysfunction
Abstract
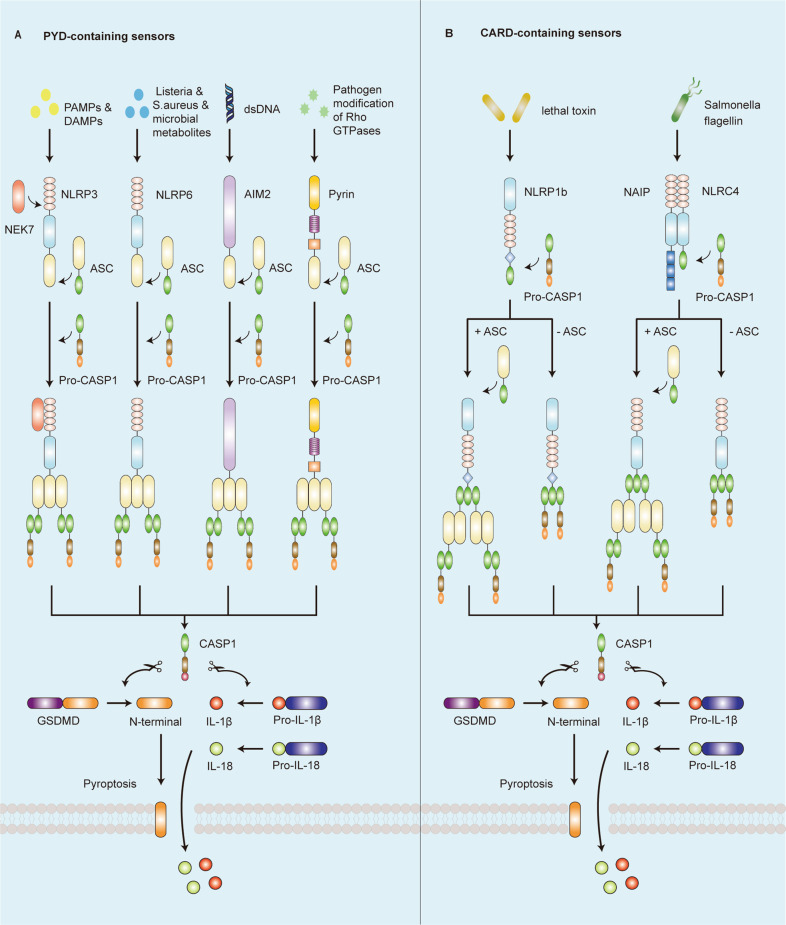
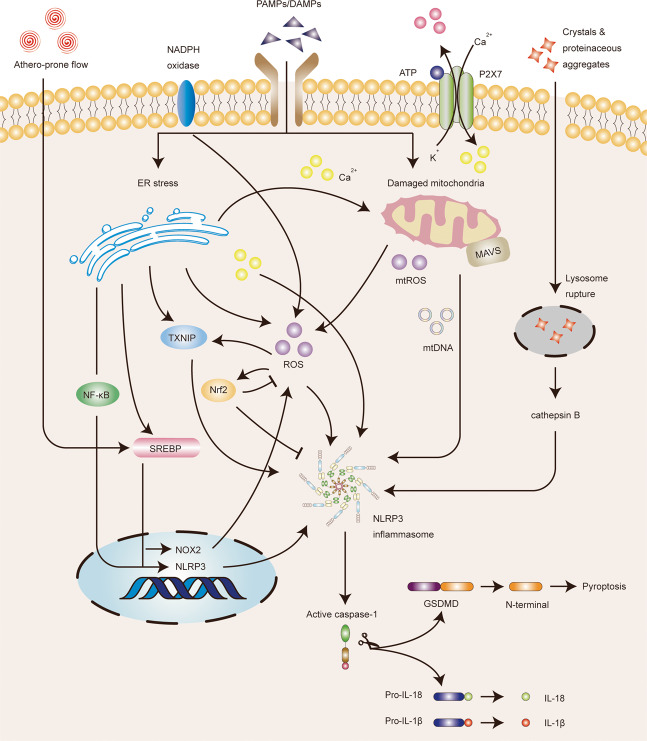
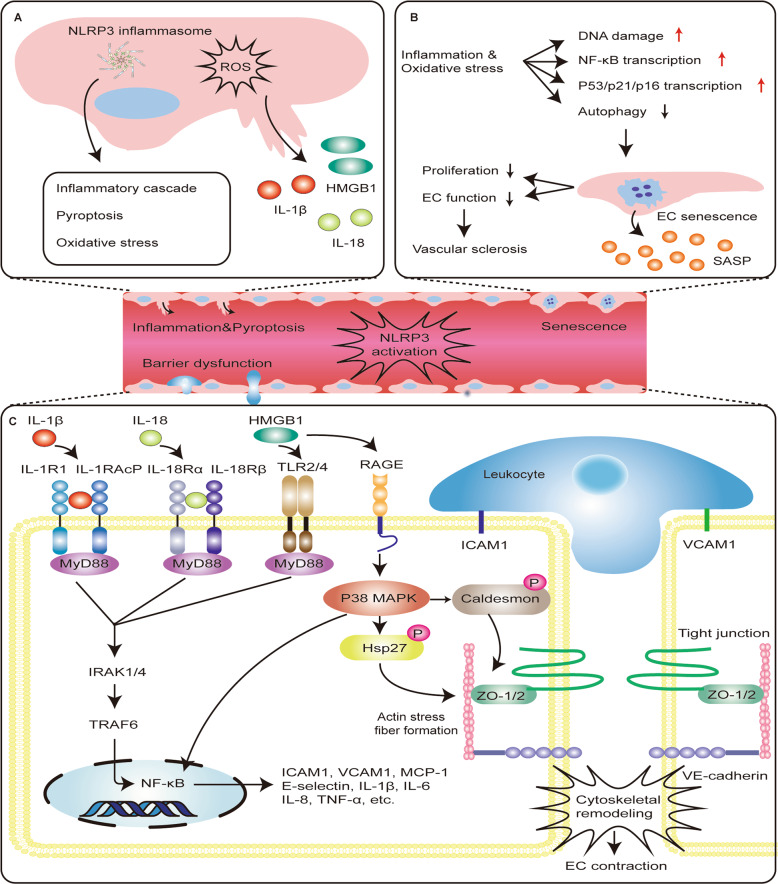
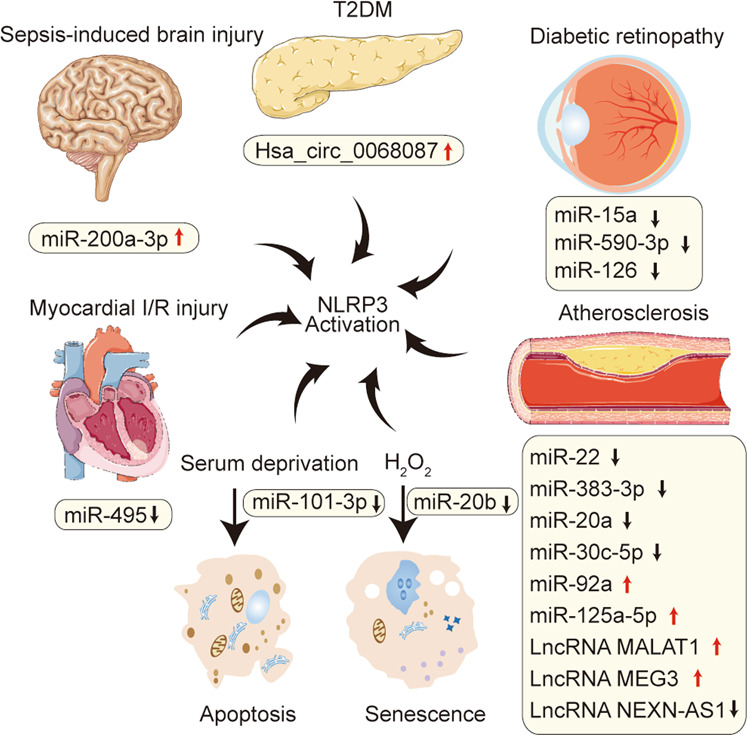
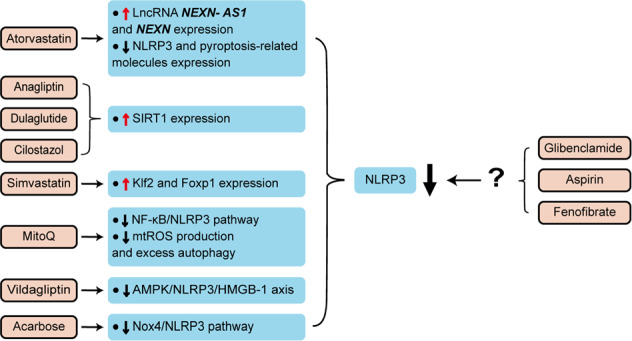
Inflammasomes are a class of cytosolic protein complexes. They act as cytosolic innate immune signal receptors to sense pathogens and initiate inflammatory responses under physiological and pathological conditions. The NLR-family pyrin domain-containing protein 3 (NLRP3) inflammasome is the most characteristic multimeric protein complex. Its activation triggers the cleavage of pro-interleukin (IL)-1β and pro-IL-18, which are mediated by caspase-1, and secretes mature forms of these mediators from cells to promote the further inflammatory process and oxidative stress. Simultaneously, cells undergo pro-inflammatory programmed cell death, termed pyroptosis. The danger signals for activating NLRP3 inflammasome are very extensive, especially reactive oxygen species (ROS), which act as an intermediate trigger to activate NLRP3 inflammasome, exacerbating subsequent inflammatory cascades and cell damage. Vascular endothelium at the site of inflammation is actively involved in the regulation of inflammation progression with important implications for cardiovascular homeostasis as a dynamically adaptable interface. Endothelial dysfunction is a hallmark and predictor for cardiovascular ailments or adverse cardiovascular events, such as coronary artery disease, diabetes mellitus, hypertension, and hypercholesterolemia. The loss of proper endothelial function may lead to tissue swelling, chronic inflammation, and the formation of thrombi. As such, elimination of endothelial cell inflammation or activation is of clinical relevance. In this review, we provided a comprehensive perspective on the pivotal role of NLRP3 inflammasome activation in aggravating oxidative stress and endothelial dysfunction and the possible underlying mechanisms. Furthermore, we highlighted the contribution of noncoding RNAs to NLRP3 inflammasome activation-associated endothelial dysfunction, and outlined potential clinical drugs targeting NLRP3 inflammasome involved in endothelial dysfunction. Collectively, this summary provides recent developments and perspectives on how NLRP3 inflammasome interferes with endothelial dysfunction and the potential research value of NLRP3 inflammasome as a potential mediator of endothelial dysfunction.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
-
- Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020;20:95–112. - PubMed
-
- Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016;16:35–50. - PubMed
-
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. - PubMed
-
- Han J, Ulevitch RJ. Limiting inflammatory responses during activation of innate immunity. Nat. Immunol. 2005;6:1198–1205. - PubMed
-
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002;10:417–426. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
