

ATXN1 repeat expansions confer risk for amyotrophic lateral sclerosis and contribute to TDP-43 mislocalization
- PMID: 32954321
- PMCID: PMC7425293
- DOI: 10.1093/braincomms/fcaa064
ATXN1 repeat expansions confer risk for amyotrophic lateral sclerosis and contribute to TDP-43 mislocalization
Abstract
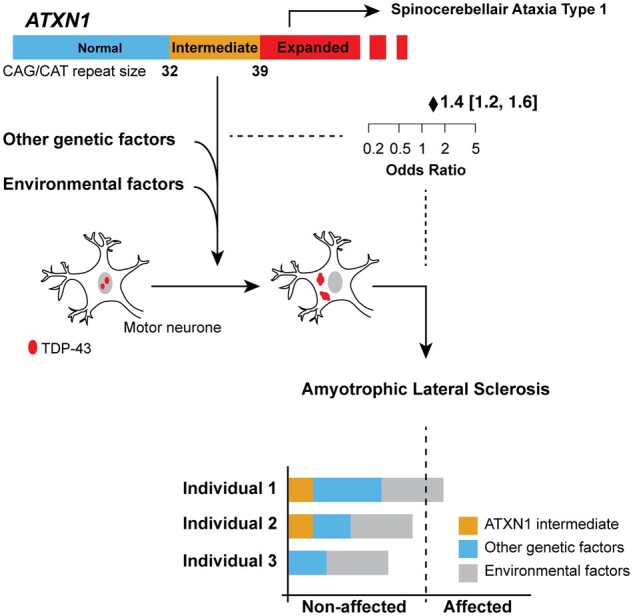
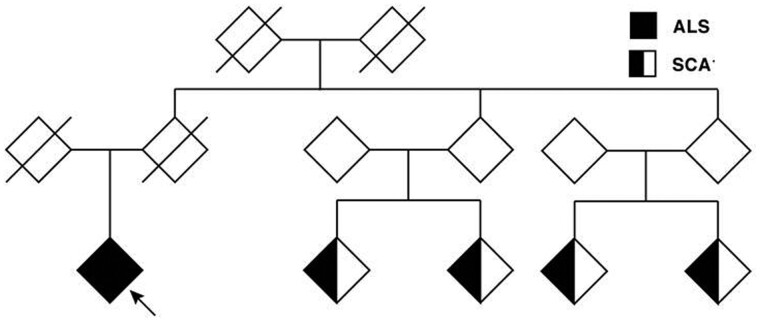
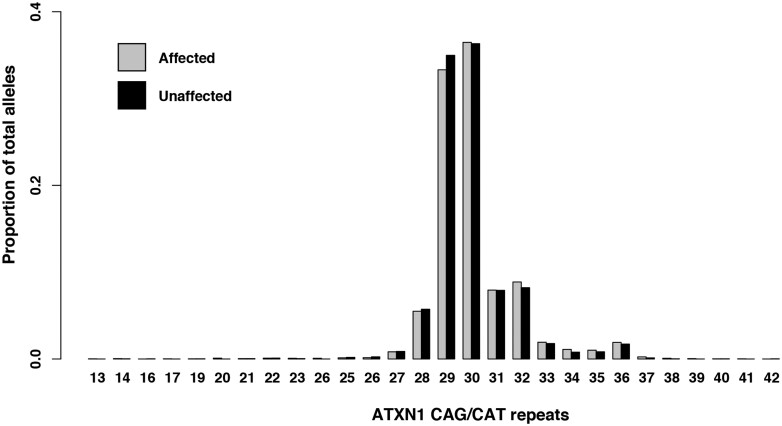
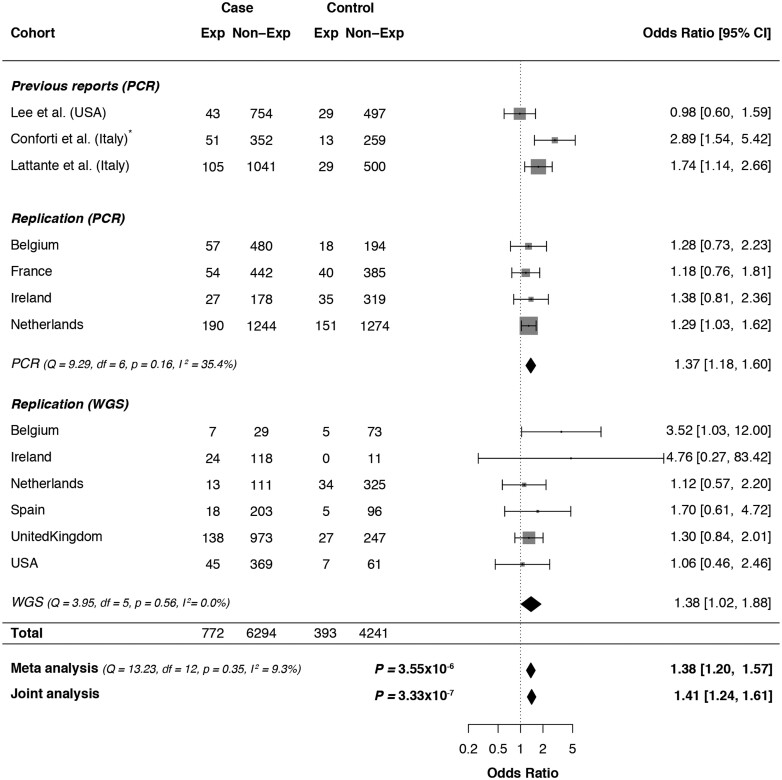
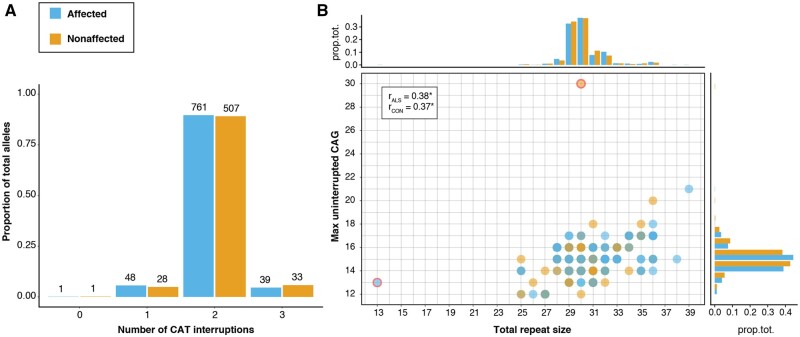
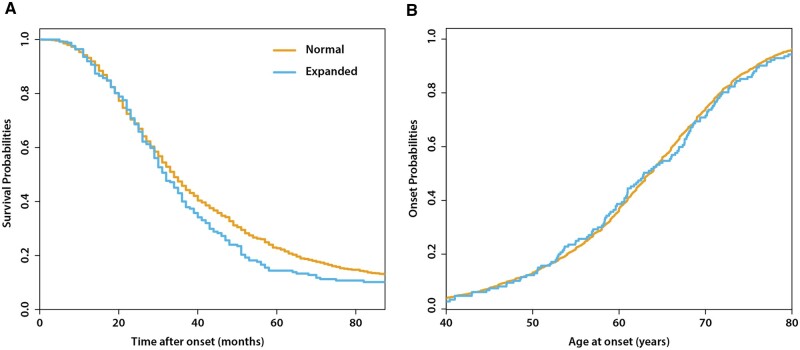
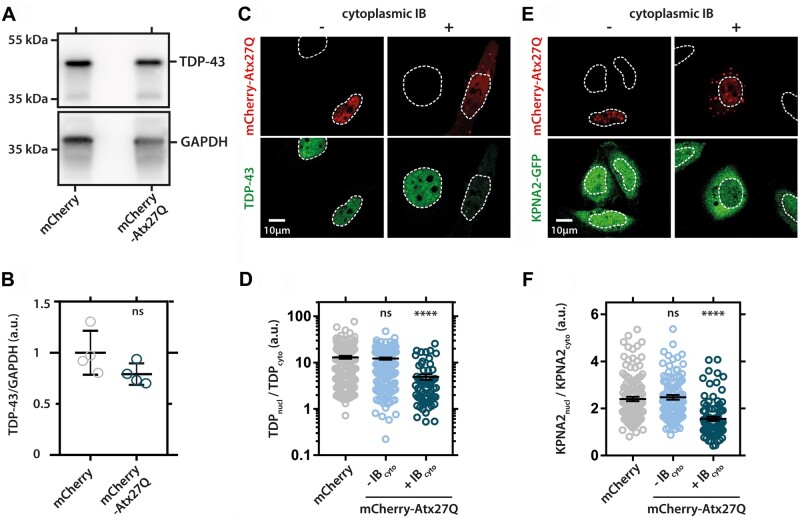
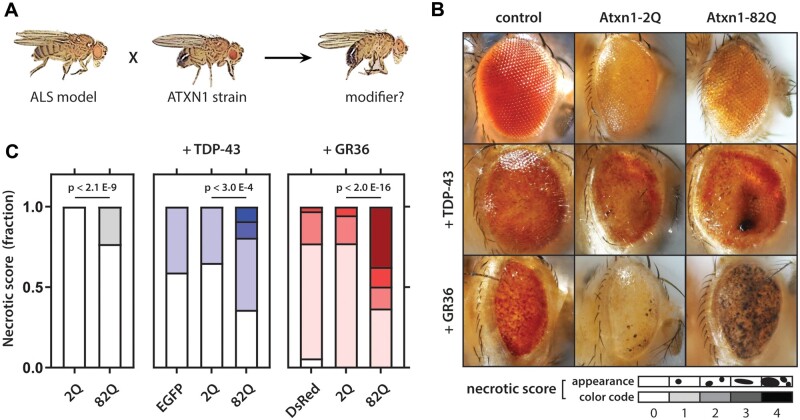
Increasingly, repeat expansions are being identified as part of the complex genetic architecture of amyotrophic lateral sclerosis. To date, several repeat expansions have been genetically associated with the disease: intronic repeat expansions in C9orf72, polyglutamine expansions in ATXN2 and polyalanine expansions in NIPA1. Together with previously published data, the identification of an amyotrophic lateral sclerosis patient with a family history of spinocerebellar ataxia type 1, caused by polyglutamine expansions in ATXN1, suggested a similar disease association for the repeat expansion in ATXN1. We, therefore, performed a large-scale international study in 11 700 individuals, in which we showed a significant association between intermediate ATXN1 repeat expansions and amyotrophic lateral sclerosis (P = 3.33 × 10-7). Subsequent functional experiments have shown that ATXN1 reduces the nucleocytoplasmic ratio of TDP-43 and enhances amyotrophic lateral sclerosis phenotypes in Drosophila, further emphasizing the role of polyglutamine repeat expansions in the pathophysiology of amyotrophic lateral sclerosis.
Keywords: DNA repeat expansion; amyotrophic lateral sclerosis; genetic association study; trinucleotide repeat expansions.
© The Author(s) (2020). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
References
-
- Al-Chalabi A, van den Berg LH, Veldink J.. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol 2017; 13: 96–104. - PubMed
-
- Andersen PM, Al-Chalabi A.. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 2011; 7: 603–15. - PubMed
-
- Banfi S, Servadio A, Chung MY, Kwiatkowski TJ Jr., McCall AE, Duvick LA, et al.Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet 1994; 7: 513–20. - PubMed
Grants and funding
- ALCHALABI-DOBSON/APR14/829-791/MNDA_/Motor Neurone Disease Association/United Kingdom
- R56 NS073873/NS/NINDS NIH HHS/United States
- MR/L501529/1/MRC_/Medical Research Council/United Kingdom
- G0600974/MRC_/Medical Research Council/United Kingdom
- AL-CHALABI/APR15/844-791/MNDA_/Motor Neurone Disease Association/United Kingdom
LinkOut - more resources
Full Text Sources
Molecular Biology Databases