

Clinical, functional and genetic characterization of 16 patients suffering from chronic granulomatous disease variants - identification of 11 novel mutations in CYBB
- PMID: 32954498
- PMCID: PMC7806450
- DOI: 10.1111/cei.13520
Clinical, functional and genetic characterization of 16 patients suffering from chronic granulomatous disease variants - identification of 11 novel mutations in CYBB
Abstract
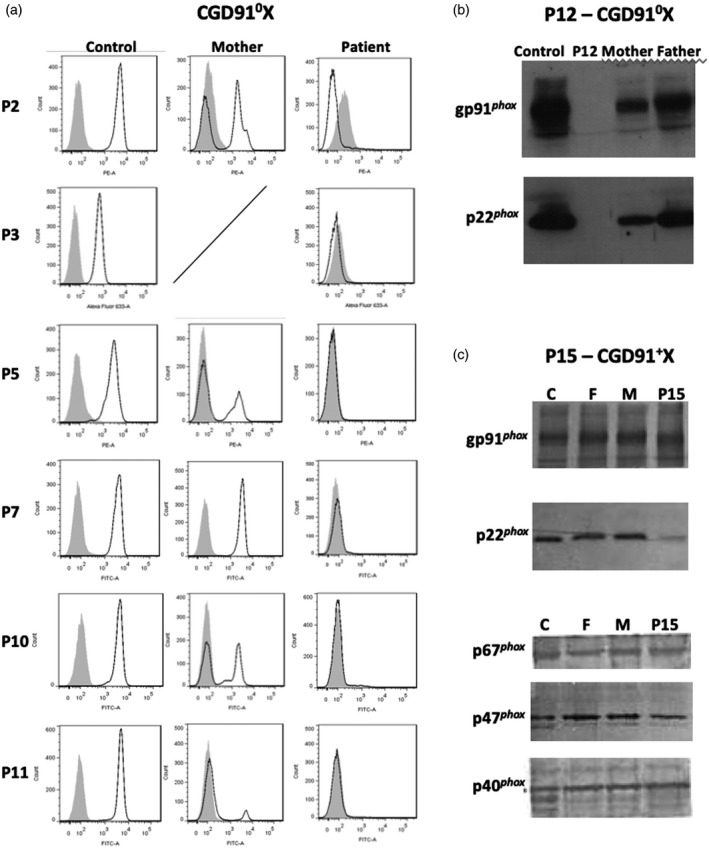
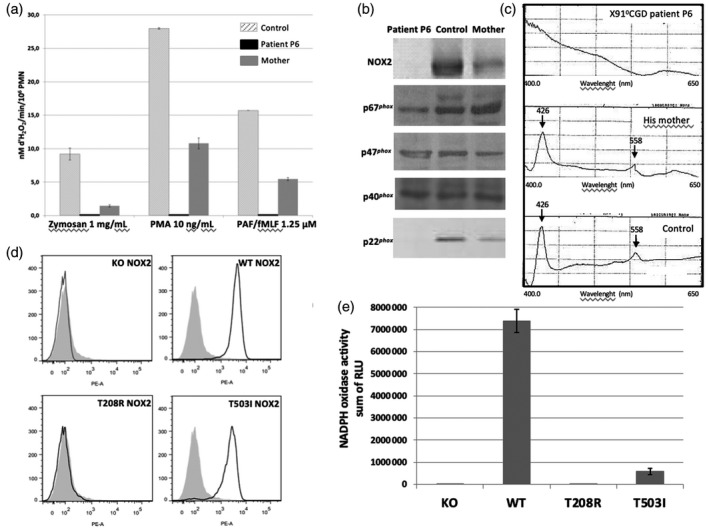
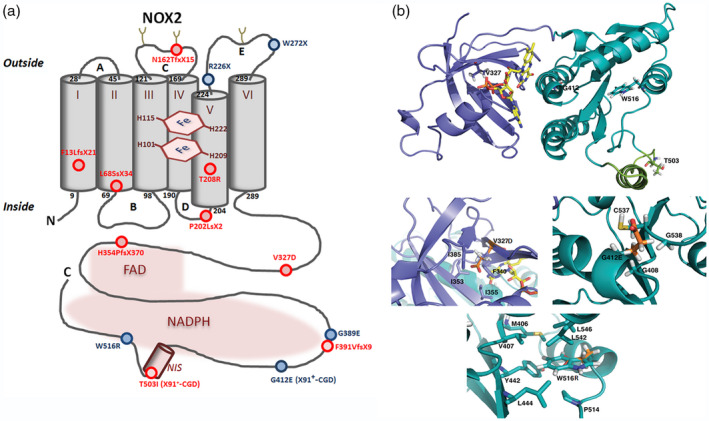
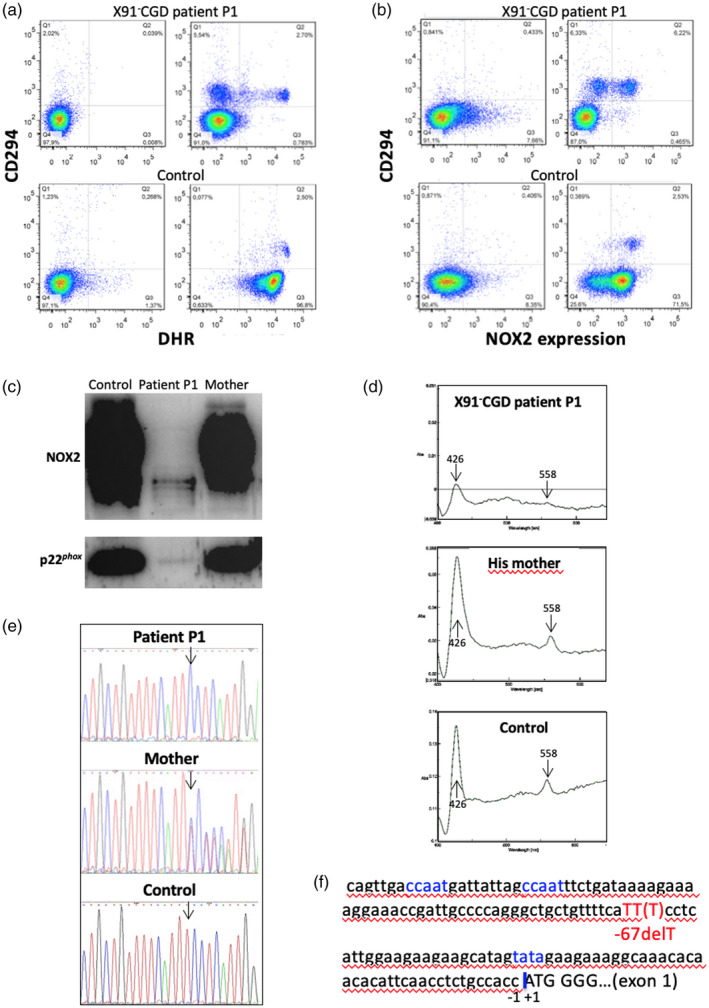
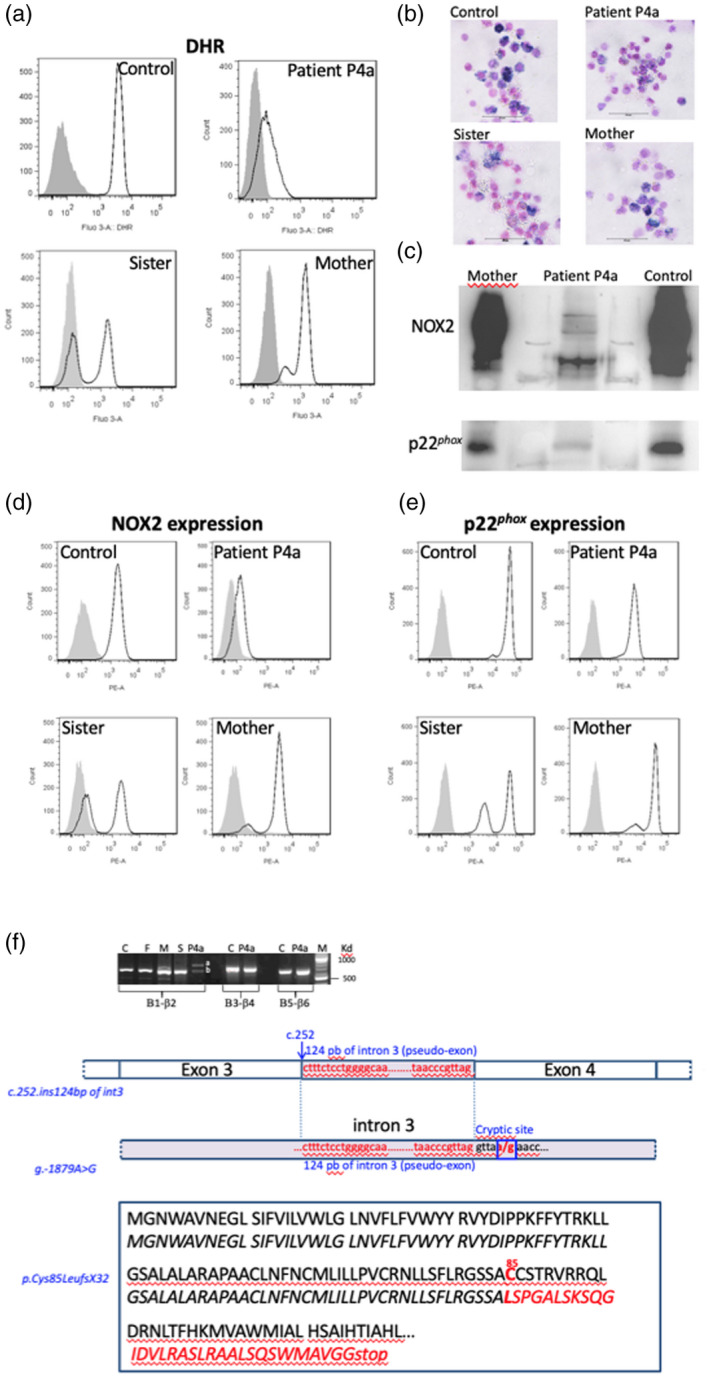
Chronic granulomatous disease (CGD) is a rare inherited disorder in which phagocytes lack nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity. The most common form is the X-linked CGD (X91-CGD), caused by mutations in the CYBB gene. Clinical, functional and genetic characterizations of 16 CGD cases of male patients and their relatives were performed. We classified them as suffering from different variants of CGD (X910 , X91- or X91+ ), according to NADPH oxidase 2 (NOX2) expression and NADPH oxidase activity in neutrophils. Eleven mutations were novel (nine X910 -CGD and two X91- -CGD). One X910 -CGD was due to a new and extremely rare double missense mutation Thr208Arg-Thr503Ile. We investigated the pathological impact of each single mutation using stable transfection of each mutated cDNA in the NOX2 knock-out PLB-985 cell line. Both mutations leading to X91- -CGD were also novel; one deletion, c.-67delT, was localized in the promoter region of CYBB; the second c.253-1879A>G mutation activates a splicing donor site, which unveils a cryptic acceptor site leading to the inclusion of a 124-nucleotide pseudo-exon between exons 3 and 4 and responsible for the partial loss of NOX2 expression. Both X91- -CGD mutations were characterized by a low cytochrome b558 expression and a faint NADPH oxidase activity. The functional impact of new missense mutations is discussed in the context of a new three-dimensional model of the dehydrogenase domain of NOX2. Our study demonstrates that low NADPH oxidase activity found in both X91- -CGD patients correlates with mild clinical forms of CGD, whereas X910 -CGD and X91+ -CGD cases remain the most clinically severe forms.
Keywords: NADPH oxidase; NOX; X-linked CGD variants; clinical severity.
© 2020 British Society for Immunology.
Conflict of interest statement
On behalf of all authors, the corresponding author states that there are no conflicts of interest.
Figures
References
-
- Mahlaoui N, Picard C, Bach P et al Genetic diagnosis of primary immunodeficiencies: a survey of the French national registry. J Allergy Clin Immunol 2019; 143:1646–1649.e1610. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
