

Comment
doi: 10.1073/pnas.2017726117.
Epub 2020 Sep 21.
Low genetic diversity may be an Achilles heel of SARS-CoV-2
Affiliations
- PMID: 32958678
- PMCID: PMC7547206
- DOI: 10.1073/pnas.2017726117
Item in Clipboard
Comment
Low genetic diversity may be an Achilles heel of SARS-CoV-2
Proc Natl Acad Sci U S A.
.
No abstract available
Conflict of interest statement
The authors declare no competing interest.
Figures
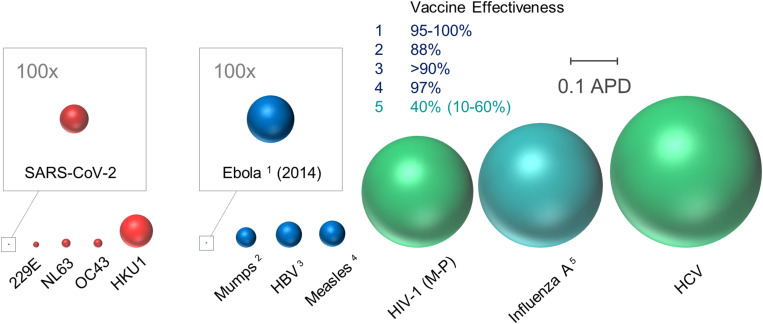
Comparative genetic diversity among coronaviruses and select viral pathogens. As indicated by the scale bar, sphere radius reflects average pairwise distances (APD) of viral surface glycoprotein gene sequences among different viruses. Diversities among coronaviruses (for which no vaccines have been developed to date) are indicated in red, and those of other viruses for which effective vaccines are available or unavailable are shown in blue and green, respectively. Since 2005, the average effectiveness of combination influenza seasonal vaccines (influenza A: H1N1, H2N3, influenza B) has been 40%. Accordingly, genetic diversity of influenza A is depicted by blue-green shading to reflect an intermediate level of vaccine effectiveness. Sequences were obtained from public databases and identical sequences were included only once. MEGA7 software was used to calculate APD among gene segments encoding proteins involved in attachment/entry: Spike or Spike-like human coronaviruses (SARS-CoV-2, 229E, NL63, OC43, and HKU1), spike glycoprotein (Ebola), HN (mumps), S (HBV), H (measles), Env (HIV-1), HA (influenza A), and E1 (HCV). More specifically, HIV-1 Group M subtypes A–D, F–H, J–K, CRF01_AE, and CRF02_AG; HBV serotypes A–H; HCV genotypes 1a–c, 2a–b, 4a, 5a, 6a, 6k, and 6m; and influenza A H1N1 pdm09, seasonal H1N1, H3N2, and H5N1 were included. Majority-rule consensus of unique sequences for HIV-1 (Group M, N, O, and P), HBV, HCV, and influenza A was performed in Seaview v4.7. Total numbers of sequences analyzed: SARS-CoV-2 (21,554), 229E (25), NL63 (52), OC43 (79), HKU1 (38), Ebola (578), mumps (341), HBV (10,271), measles (38), HIV-1 (5,603), influenza A (133), and HCV (439).
Comment on
-
A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants.Proc Natl Acad Sci U S A. 2020 Sep 22;117(38):23652-23662. doi: 10.1073/pnas.2008281117. Epub 2020 Aug 31. Proc Natl Acad Sci U S A. 2020. PMID: 32868447 Free PMC article.
References
-
- Duffy S., Shackelton L. A., Holmes E. C., Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 9, 267–276 (2008). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
